
Abstract
Objective:
Muscarinic receptor dysfunction has been suggested to play an important role in the pathophysiology of schizophrenia. Recently, it has also become clear that immune reactivity directed against neurotransmitter receptors may play a pathogenic role in some cases of schizophrenia. The aim of this review is to summarize the case for muscarinic receptor dysfunction in schizophrenia and the evidence supporting the hypothesis that this dysfunction is related to the development of muscarinic receptor–targeting antibodies.
Method:
The article reviews studies of muscarinic receptors and the presence and potential role(s) of anti-muscarinic acetylcholine receptor antibodies in people with schizophrenia.
Results:
There is accumulating evidence that altered or deficient muscarinic signalling underlies some of the key clinical features of schizophrenia. Although the number of studies investigating anti-muscarinic acetylcholine receptor antibodies in schizophrenia is relatively small, they consistently demonstrate that such antibodies are present in a proportion of patients. This evidence suggests that these antibodies could have pathogenic effects or exist as a biomarker to an unknown pathophysiological process in schizophrenia.
Conclusion:
The presence of elevated levels of anti-muscarinic acetylcholine receptor antibodies may identify a subgroup of people with schizophrenia, potentially informing aetiopathogenesis, clinical presentation and treatment. To date, all studies have examined antibodies in participants with chronic schizophrenia, who have likely received antipsychotic medication for many years. As these medications modulate immune functions and regulate receptor densities, it is recommended that future studies screen for the presence of anti-muscarinic antibodies in people experiencing their first episode of psychosis.
Introduction
Schizophrenia is a persistent heterogeneous syndrome with a life-time prevalence of almost 1% (Saha et al., 2005) and is associated with high levels of disability (Charlson et al., 2018). The aetiology remains elusive: the predominant hypothesis asserts that schizophrenia is a neurodevelopmental disorder with a strong genetic predisposition (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) interacting with environmental risk (Stilo et al., 2011). However, converging evidence implicates the immune system in the emergence of schizophrenia in some individuals. Increased levels of circulating pro-inflammatory cytokines (Goldsmith et al., 2016; Miller et al., 2011) and aberrations in lymphocyte levels and ratios (Delaney, 2016; Miller et al., 2013) have been reported in people with schizophrenia. Many of these observed alterations in immune parameters are common to other autoimmune diseases, suggesting that some people with schizophrenia could have an autoimmune basis for their disease (Jones et al., 2005).
Furthermore, genome-wide association studies (GWAS) of people with schizophrenia have repeatedly found significant associations in the region encoding the major histocompatibility complex (MHC; Kodavali et al., 2014), which spans several megabases of chromosome 6p and encompasses many genes relating to immune function (Shiina et al., 2009). The strongest association within the MHC has been mapped to the complement component 4 (C4) genes (Sekar et al., 2016), which are part of the complement cascade that plays important roles in the innate immune response and which is necessary for many effector functions mediated by pathogenic antibodies. There are also numerous reports of elevated levels of autoantibodies that target brain components in people with schizophrenia [reviewed by Hoffmann et al. (2016) and Jones et al. (2005)]. Some of these autoantibodies target cell surface molecules on neurons. One of the most prominent targets in this regard is the N-methyl-D-aspartate receptor (NMDAR) (Lennox et al., 2017; Scott et al., 2018; Steiner et al., 2015; Zandi et al., 2014). Three studies on anti-NMDAR antibodies in patients with first episode psychosis have reported positive clinical outcomes of immunosuppressive therapy on both the levels of anti-NMDAR antibodies and disease symptoms (Lennox et al., 2019; Scott et al., 2018; Zandi et al., 2014), suggesting these antibodies may be pathogenic in some patients.
It is likely that autoantibodies targeting other neurotransmitter receptors also exist and could be pathogenic in patients with psychotic symptoms. One potentially interesting group of molecules are the muscarinic acetylcholine receptors (mAChRs) (abbreviated throughout as CHRM). Elevated levels of anti-CHRM antibodies have been reported in up to one-third of people living with schizophrenia (Jones et al., 2014; Tanaka et al., 2003), and their presence has been reported to correlate with negative symptoms (Jones et al., 2014). As such, a better understanding of immune-regulated dysfunction in the muscarinic system may help to inform the aetiology and treatment options of schizophrenia in some individuals. This review aims to synthesize and critically appraise the literature regarding CHRM in schizophrenia, and the role that anti-CHRM autoantibodies might play in the pathogenesis of schizophrenia.
Acetylcholine signalling and the mAChRs
In the central nervous system (CNS), acetylcholine-mediated neurotransmission is essential in higher cognitive processes such as learning, attention and memory (Newman et al., 2012) and in helping to coordinate the activity of neural networks that underlie complex behaviours (Picciotto et al., 2012). Cholinergic neurons form an intricate brain-wide network consisting of three main components (Scarr et al., 2013b). Cholinergic projections from the basal forebrain nuclei innervate most cortical regions, the hippocampus and some subcortical regions. In addition, cholinergic projections from the pedunculopontine and laterodorsal tegmental nuclei in the brainstem innervate the thalamus, midbrain and other brainstem regions. Finally, an intrinsic network of cholinergic interneurons are found in the striatum (Scarr et al., 2013b). In addition to the spatial location of cholinergic neurons, acetylcholine signalling is highly dependent on the expression pattern of associated receptors and receptor subtypes.
Acetylcholine receptors consist of two major subtypes: nicotinic acetylcholine receptors (CHRN) and CHRM. The CHRN are a group of ligand-gated ion channel receptors, otherwise known as ionotropic receptors, that are excitatory, located both pre- and post-synaptically in the CNS, and modulate the release of various neurotransmitters (Dani and Bertrand, 2007). Dysfunction of the nicotinic cholinergic system is implicated in the neuropathology of schizophrenia, Parkinson’s disease, Alzheimer’s, dementia and addiction. The CHRN have previously been comprehensively reviewed (Albuquerque et al., 2009).
Apart from binding acetylcholine, CHRM have little in common in structure or function with the CHRN. CHRM are part of the superfamily of G protein–coupled receptors (GPCRs), otherwise known as metabotropic receptors (Caulfield, 1993). When a ligand binds to a GPCR, it activates and releases the coupled G protein, which, depending on the type of G protein, signals a cascade of primary effector proteins and secondary messengers. The CHRMs consist of five subtypes, M1-5 (Caulfield and Birdsall, 1998), which can be further categorized into two groups. The first group, referred to as ‘M1-like’ receptors, consist of the M1, M3 and M5 subtypes. M1-like receptors are located post-synaptically and are coupled with Gq/G11 proteins, which have excitatory downstream effects (Jones et al., 2012). The second group, the ‘M2-like’ receptors (M2 and M4 subtypes), are located pre- and post-synaptically and are coupled with Gi/Go proteins which have predominantly inhibitory effects (Figure 1[A]).
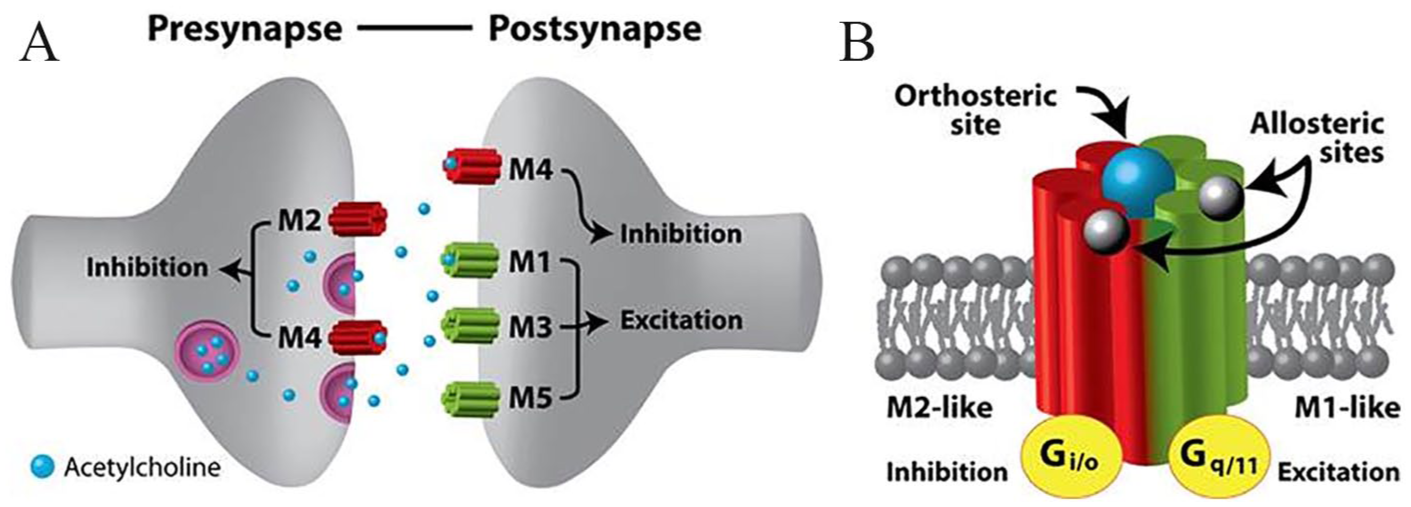
(A) Synaptic location of the CHRM. CHRM can be broadly divided into the M1-like receptors (M1, M3 and M5 subtypes), which are located post-synaptically and have excitatory downstream effects, and the M2-like receptors, which are located pre- and post-synaptically and have predominantly inhibitory effects. (B) Acetylcholine binds to the main orthosteric binding site, but its binding can be modulated via effects induced by binding of as yet unidentified ligands to allosteric binding sites located away from the orthosteric binding sites. Signals induced via binding of acetylcholine cause excitatory or inhibitory downstream effects via release of specific G proteins.
CHRMs are widely expressed, but the relative proportions of each subtype vary. The M1, M2 and M4 receptor subtypes make up the majority of CHRMs found in the mammalian CNS (Levey et al., 1991). The M1 receptor is highly expressed throughout all layers of the cerebral cortex, hippocampus and striatum, with low levels of expression in the thalamus and hindbrain (Levey et al., 1995; Levey et al., 1991; Scarr et al., 2016). The M2 receptor is also found in these regions and is the predominant CHRM in the thalamus and brainstem (Levey et al., 1991). M3 receptors are distributed in a similar fashion to the M1, albeit at considerably lower levels (Levey et al., 1994; Wolfe and Yasuda, 1995). M4 receptors are enriched in the striatum, are moderately expressed in the cerebral cortex, thalamus and hippocampus, with low levels of expression in hindbrain areas (Hersch et al., 1994; Levey et al., 1991; Wolfe and Yasuda, 1995). The M5 receptor is the subtype with the lowest levels of expression but is detectable in the hippocampus, striatum and midbrain (Wolfe and Yasuda, 1995; Yasuda et al., 1993). While CHRMs are found primarily on neurons, they are also present on other CNS cell types, including astrocytes, oligodendrocytes and microglia (De Angelis et al., 2012; Elhusseiny et al., 1999; Pannell et al., 2016). Although there has been some controversy over the detection methods used in these formative studies (Jositsch et al., 2009), localization of CHRM appears to be consistent between well-categorized immunohistochemical, labelled ligand, and mRNA expression methodologies (GTEx Consortium, 2013; Lebois et al., 2018; Piggott et al., 2002).
The capacity of the acetylcholine binding site (orthosteric binding site) of CHRMs to bind to acetylcholine is regulated by binding of a ligand to another site on the CHRMs known as the allosteric binding site (Figure 1[B]). When a ligand binds to the allosteric site, it induces a conformational change in the receptor, which can increase or decrease the binding affinity of the orthosteric site to acetylcholine. Although ‘natural’ allosteric ligands in the brain have not yet been identified, several synthetic compounds with varying affinities for the allosteric binding sites have been produced. There is high sequence homology at the orthosteric binding site across all of the CHRM subtypes (M1–M5). In contrast, the allosteric binding sites have large subtype-specific variations in binding affinity to allosteric agents (Gregory et al., 2007). Therefore, these allosteric binding sites are essential to understanding the function of the CHRMs in the brain, and their involvement in schizophrenia and other CNS disorders (Conn et al., 2009).
Schizophrenia
People with schizophrenia commonly experience positive (e.g. psychotic symptoms such as delusions and hallucinations), negative (e.g. blunted affect, social withdrawal and apathy) and cognitive (impairment in memory, attention and processing speed) symptoms. Ultimately, in the absence of reliable biomarkers or supportive investigations, the diagnosis of schizophrenia is reliant on a phenotype of heterogeneous self-reported and observable behavioural symptoms. Considering this heterogeneity in clinical presentation, it is likely that a multitude of pathologies account for the syndrome (Dutta et al., 2007; Fischer and Carpenter, 2009).
Multiple genetic, neurodevelopmental, environmental, and social studies suggest that striatal hyperdopaminergia is responsible for the positive symptoms of schizophrenia in most cases [reviewed by Howes and Kapur (2009) and Kesby et al. (2018)]. As such, current antipsychotic pharmacotherapy, which primarily involves dopamine receptor D2 (DRD2) antagonism, has greatest effectiveness in the management of positive symptoms. However, the aetiologies of cognitive dysfunction and negative symptoms are less clear, and current antipsychotic agents targeting DRD2 receptors show negligible efficacy in treating negative symptoms and alleviating cognitive dysfunction (Minzenberg and Carter, 2012; Swartz et al., 2008). The full spectrum of schizophrenia likely involves multiple neurotransmitter systems other than dopamine, including glutamate, gamma-amino butyric acid (GABA), serotonin, and acetylcholine (Scarr et al., 2013b). Since acetylcholine is a neuromodulator with neural projections coincident to dopaminergic and other neurotransmitter systems implicated in schizophrenia, alterations to CHRM signalling could play an aetiopathological role.
Modulation of the cholinergic system in schizophrenia
Prior to modern antipsychotic therapy, agents targeting the cholinergic system were used in the treatment of schizophrenia. However, compared with the DRD2 antagonists, they caused a high burden of side effects, limiting further investigation of their clinical utility. There are, however, several indications from the literature that acetylcholine levels are important in negative and cognitive symptoms of schizophrenia. A Cochrane review of adjunct use of acetylcholinesterase inhibitors (compounds which increase the availability of acetylcholine) in people living with schizophrenia reported improvements in general psychopathology, negative symptomatology and depressive symptoms, and in cognitive domains of memory and attention (Singh et al., 2012). Consistent with this, a systematic review (Desmarais et al., 2012) identified three studies showing significant improvements in cognitive functioning following discontinuation of anticholinergic medication (which decrease the availability of acetylcholine) in schizophrenia patients receiving antipsychotics. Furthermore, levels of anticholinergic activity in a cohort of people with schizophrenia was negatively correlated with memory and global cognition, accounting for 20% of the variance in global cognition change score, independent of the effects of intelligence quotient (IQ), age and symptom severity (Vinogradov et al., 2009). While these studies suggest modulation of the cholinergic system can influence schizophrenia symptomatology, they do not clarify the role of muscarinic and nicotinic systems, nor the relative importance of muscarinic subtypes.
Studies investigating a 2-day trial of biperiden hydrochloride (a CHRM antagonist with some selectivity for the M1) in unmedicated people with schizophrenia had differential effects on symptomatology, increasing positive symptoms but reducing negative symptoms (Tandon et al., 1992; Tandon et al., 1991). Furthermore, a trial using xanomeline, a relatively selective M1 and M4 agonist, demonstrated improvement in symptoms and cognitive function relative to placebo (Shekhar et al., 2008). In this trial, 20 people who were either experiencing an acute exacerbation of schizophrenia or responding poorly to antipsychotic therapy were randomly assigned to increasing doses of xanomeline or placebo over 4 weeks. The xanomeline group experienced significant decreases in mean total scores in both the Brief Psychiatric Rating Scale (Overall and Gorham, 1962) and Positive and Negative Syndrome Scale (PANSS; Kay et al., 1987). In the cognitive test battery, the xanomeline group had robust improvements in measures of verbal learning and short-term memory function, although there were no significant improvements in attention or speed of information processing (Shekhar et al., 2008). While xanomeline showed promise in treating schizophrenia symptoms, an increase in adverse gastrointestinal effects caused by muscarinic activation of the peripheral nervous system has hindered further research. Currently, a phase II clinical trial (NCT03697252) is being conducted using a co-formulation of xanomeline and trospium known as KarXT, for the treatment of acute exacerbation of schizophrenia. Trospium is a peripheral M2 and M3 receptor antagonist, which has been co-formulated with xanomeline to reduce peripheral adverse effects. Results of initial phase I testing of KarXT have not yet been published, although material released by the manufacturer of the drug states that it demonstrated tolerability at dose levels exceeding those shown to be efficacious in pervious xanomeline studies.
As discussed previously, CHRM have allosteric sites distinct from the main acetylcholine-binding site. The key advantage of targeting allosteric binding sites is their low degree of sequence homology across the various muscarinic subtypes (M1–M5). Agents that target the main orthosteric sites (which have high sequence homology) induce effects on multiple muscarinic subtypes, whereas allosteric agents have the potential to allow CHRM subtype-specific targeting. Of particular interest are allosteric agonists and positive allosteric modulators (PAMs) with high specificity for the M1 and M4 receptor subtypes. These compounds, when tested in animal models of schizophrenia, have produced encouraging results [reviewed by Carruthers et al. (2015) and Foster and Conn (2017)]. Critically, M1 PAMs appear to have potential to reduce cognitive and negative symptoms associated with changes in cortical plasticity. M4 PAMs have shown antipsychotic-like effects that are thought to be driven by a reduction in dopaminergic signalling. The development of pharmacotherapy using PAMs and allosteric agonists to target muscarinic subtypes may offer therapeutic benefit, with reduced cholinergic side effects.
Muscarinic receptors in schizophrenia
Reductions in CHRM densities have been repeatedly observed in neuropathological post-mortem studies of brains from people with schizophrenia. In the cortex, M1 receptor density reductions are evident (Dean et al., 2002; Gibbons et al., 2013; Mancama et al., 2003; Seo et al., 2014), while M2, M3 and M4 receptor densities remain unchanged (Dean et al., 2002; Scarr et al., 2006; Seo et al., 2014; Zavitsanou et al., 2005). Furthermore, reduced density of M1 receptors in the cortex appears to be specific to schizophrenia, with no reductions observed in bipolar affective disorder or major depressive disorder (Zavitsanou et al., 2004). In contrast, in subcortical regions, there is decreased M4 receptor density observed in the hippocampus (Scarr et al., 2007) and reductions in M1, M4 and possibly M2 subtypes in striatal regions (Crook et al., 1999; Dean et al., 2000; Dean et al., 2015).
In post-mortem studies, investigating the density of CHRM in the dorsolateral prefrontal cortex (Brodmann area 9), approximately 25% of schizophrenia samples (compared to both other schizophrenia samples and controls) show a pronounced (>70%) reduction of the binding of the muscarinic agonist [3H] pirenzepine (Scarr et al., 2009). The authors referred to these patients as having muscarinic receptor deficit schizophrenia (MRDS). Apart from pirenzepine binding, the MRDS subgroup was not able to be distinguished from the remaining schizophrenia cohort based on the CHRM1 genotype, gender, age, duration of illness, pharmacotherapy, or cause of death (Scarr et al., 2009). Further investigations have found that MRDS cases also have profound, widespread losses in M1 receptor densities across other cortical regions (namely Brodmann areas 10, 46, 44 and 24) in comparison with the remaining schizophrenia cohort (Gibbons et al., 2013). Extending beyond the cortex, MRDS cases also have a reduction, compared to controls and other schizophrenia patients, in M1 as well as M2 and/or M4 receptors in the striatum (Dean et al., 2015). It is currently unknown whether the clinical presentation of patients with MRDS differs from non-MRDS patients. However, if the neuropathological findings translate to people living with schizophrenia, the pronounced reduction in M1 receptor densities would be expected to have significant functional consequences (Scarr and Dean, 2008). Intriguingly, the post-mortem studies indicated that while there is pronounced loss of M1 receptors in the MRDS subtype, the residual M1 population has increased receptor–G protein coupling efficiency, suggesting an adaptive change to compensate for receptor paucity (Salah-Uddin et al., 2009). In addition, the response to treatment with benzyl quinolone carboxylic acid (BQCA), a selective M1 PAM, is decreased in MRDS (Dean et al., 2016).
In an attempt to identify the cause of M1 receptor reductions in MRDS, specific markers of gene expression were investigated (Scarr et al., 2013a; Scarr et al., 2018b). Increased cortical levels of microRNA (miR)-107 were found in MRDS in comparison to controls. MiR-107 is thought to regulate the expression of M1 receptors and may contribute to the marked loss of this specific receptor subtype (Scarr et al., 2013a). Furthermore, levels of mRNA for 65 genes that regulate cellular movement and cell-to-cell signalling were significantly different in the cortex of MRDS compared with non-MRDS subjects. It was suggested that the identified mRNA changes argue for a prominent role of glial cells in MRDS (Scarr et al., 2018b). However, the aetiology of MRDS remains unknown.
A number of studies have investigated central CHRM availability in vivo in schizophrenia using single photon emission computed tomography (SPECT). The first of these used [123I]-iodoquinuclidinyl benzilate, which binds equally to all CHRM subtypes, in 12 unmedicated people with schizophrenia and 10 age- and gender-matched healthy controls (Raedler et al., 2003). The authors reported significant reductions in CHRM availability, up to 33% less than controls, in schizophrenia-relevant brain regions. Decreased CHRM availability in both the frontal cortex and striatum was also correlated with increased positive symptom subscale scores on the PANSS (Raedler et al., 2003). More recently, a similar study has used [123I]-iododexetimide, which has a high binding affinity for M1 and M4 receptor subtypes, to investigate the relationship between the availability of these receptors, clinical symptoms and cognitive functioning in 12 patients with schizophrenia (Bakker et al., 2018). Although inferences cannot be made on an overall reduction of CHRM in the schizophrenia population from this study, as a control group was not included, it is novel in its observations on the relationship between CHRM availability and clinical symptoms. This study reported that decreased receptor availability in the dorsolateral prefrontal cortex was associated with increased severity and presence of negative symptoms measured by the PANSS, and with lower verbal learning and memory. Since the M1 receptor is the predominant CHRM expressed in the prefrontal cortex, it was concluded that this relationship was due to decreased M1 receptor availability. In contrast, decreased hippocampal M1/M4 receptor availability was associated with worse delayed recognition of verbal information. Overall, the above studies (which are summarized in Figure 2) suggest that changes in the levels or availability of CHRM are associated with symptom presentation in schizophrenia, although further studies in larger numbers of individuals will be required to define the exact relationships.
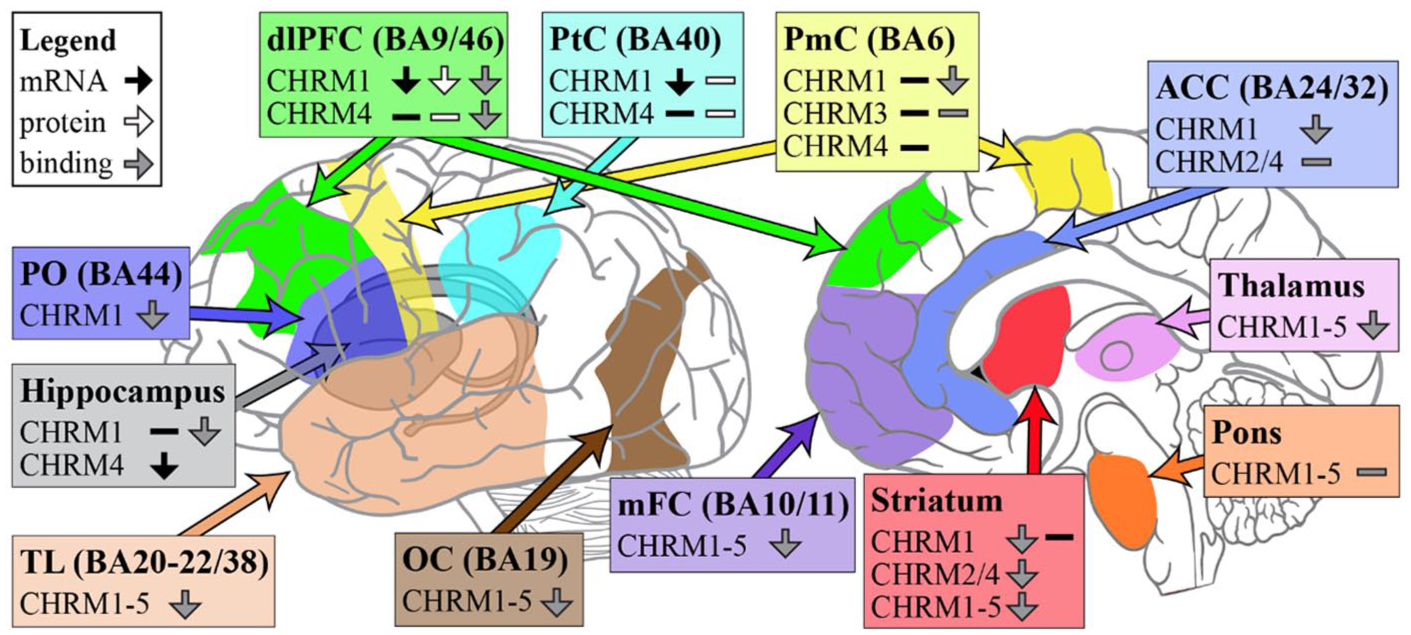
Summary of changes in RNA expression, protein levels and radioligand binding of CHRMs in people with schizophrenia compared to controls.
Autoantibodies targeting muscarinic receptors in schizophrenia
To our knowledge, only four studies have investigated muscarinic antibodies in patients with schizophrenia. These are summarized in Table 1 and described in further detail below.
Summary of studies investigating autoantibodies targeting muscarinic receptors in schizophrenia.
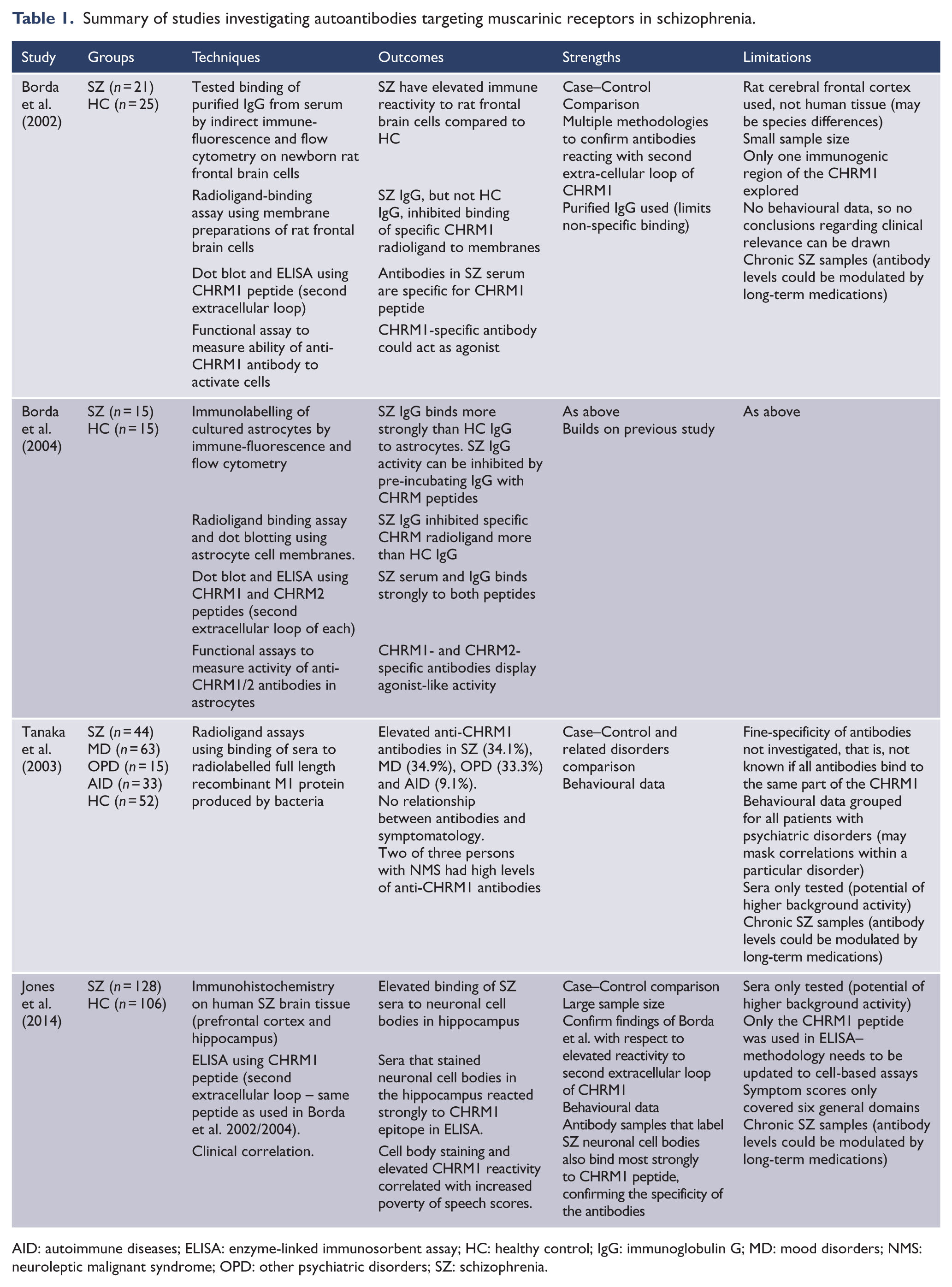
AID: autoimmune diseases; ELISA: enzyme-linked immunosorbent assay; HC: healthy control; IgG: immunoglobulin G; MD: mood disorders; NMS: neuroleptic malignant syndrome; OPD: other psychiatric disorders; SZ: schizophrenia.
The first study compared sera and purified IgG from 21 patients with schizophrenia and 25 age-matched healthy controls (Borda et al., 2002). It was shown by both indirect immunofluorescence and flow cytometry that a higher proportion of people with schizophrenia than healthy controls (14% vs 5%) had IgG in their serum that reacted with neural cells prepared from rat cortex. Further investigation showed the IgG binding to the neural cells was specific for M1 receptors. Interestingly, the anti-M1 receptor antibodies induced functional effects (changes in cGMP production, inositol phosphate accumulation and protein kinase C translocation) in rat cerebral frontal cortex that could be blocked with pirenzepine treatment (Borda et al., 2002).
In a follow-up study, the same group showed that IgG from patients with schizophrenia, but not healthy individuals, could also bind to rat astrocyte membranes. Radioligand competition assays and immunoaffinity removal of CHRM-specific antibodies demonstrated that the antibodies binding to the astrocytes were specific for the M1 and M2 receptors (Borda et al., 2004). Furthermore, these antibodies were able to induce molecular mechanisms indicative of agonist-like activity in rat astrocytes. Such agonist-like activity could potentially lead to broad cascading glial reactions, such as an increase in intracellular-free Ca2+, which triggers astrocytes to release dopamine and glutamate, both of which have a role in schizophrenia. In addition, the authors observed a decrease in cyclic adenosine monophosphate (cAMP), which plays a role in preventing oxidative neuronal damage and depressing microglial activation (Schubert et al., 2000). Critically, saturation-binding studies confirmed that schizophrenia IgG irreversibly bound to CHRMs (Borda et al., 2004). The strong avidity of these antibodies to CHRMs may help explain the reduced CHRM densities in schizophrenia and MRDS.
Another study (Tanaka et al., 2003) investigated the prevalence of anti-M1 receptor antibodies in people diagnosed with schizophrenia (n = 44), mood disorders (n = 63), ‘other’ psychiatric disorders (n = 15), or autoimmune disease (n = 33), and in healthy subjects (n=52). Using radioligand binding assays, a significantly higher frequency of patients from the groups with mental disorders had levels of anti-M1 receptor antibodies greater than the mean + 2 standard deviations of the healthy controls. This suggests that M1 receptor antibodies are more common in those with mental illness but lack specificity to schizophrenia (although the fine specificity of these anti-M1 receptor antibodies was not tested and could potentially differ between groups). The reason for the increased prevalence of anti-M1 receptor antibodies in the mental disorders studied is unclear but suggests they could be involved in the upstream processes that result in the phenotypic mood symptoms, distress and/or impairment in reality that can occur in people with mental illness. The authors did not find any significant correlations between the presence of anti-M1 receptor antibodies and symptoms, as measured by the Brief Psychiatric Rating Scale, although in this analysis, all patients with psychiatric disorders were treated as a single group, which may mask effects specific to a particular disorder (Tanaka et al., 2003). It must be noted that despite high levels of anti-CHRM1 antibodies being found in patients with mood disorders, as previously mentioned, cortical CHRM1 receptors levels are not reduced in these syndromes (Zavitsanou et al., 2004). The reason for this is unable to be determined from this study, as more would need to be known about the exact specificity of the antibodies and their isotype in order to determine if the antibodies identified in patients with mood disorders actually have the potential to produce any functional effects. Alternatively, there could be a subgroup within mood disorders that have reduced cortical CHRM1 that do not reach significance when investigating loss of CHRM in mood disorders.
In the most recent investigation of anti-M1 receptor antibodies in schizophrenia (Jones et al., 2014), sera from 128 patients with schizophrenia and 106 healthy controls were screened by enzyme-linked immunosorbent assay (ELISA) for reactivity against the same peptide of the second extracellular loop of the M1 receptor as used by Borda et al., (2004, 2002). Almost 22% of sera from people with schizophrenia showed reactivity greater than 95% of the control sera. In addition, all of the sera that showed increased anti-M1 receptor antibodies in the ELISA also labelled neuronal cell bodies in sections of hippocampal tissue from post-mortem brains of people with schizophrenia (Jones et al., 2014). The relationship between elevated levels of these antibodies and symptoms was assessed on three domains: positive symptoms (hallucinations and delusions), negative symptoms (blunted effect and speech poverty) and disorganized symptoms (disordered thought and bizarre behaviour). The presence of elevated levels of these antibodies was significantly associated with increased poverty of speech scores. This study suggests that the presence of anti-M1 receptor antibodies may be related to schizophrenia symptomatology.
Overall, the reviewed studies demonstrate that elevated levels of muscarinic antibodies are evident in schizophrenia. Furthermore, their role in the pathogenesis and symptomatology of this syndrome, and whether they are specific to schizophrenia, warrants further investigation.
Potential functional (pathogenic) effects of antibodies targeting muscarinic receptors in schizophrenia
In disorders affecting the nervous system, autoantibodies can exert pathogenic effects via a wide range of mechanisms, including antibody-mediated cell lysis, opsonisation of target proteins for attack by macrophages, crosslinking of Fc receptors, blocking or removing receptors involved in neurotransmission or cellular homeostasis or blocking of repair mechanisms (Beasley and Greer, 2015). It is possible for more than one of these mechanisms to be active in the same disease. However, there are a number of points that need to be considered when assessing the potential functional role of anti-CHRM1 antibodies. First is the specificity of the antibodies, that is, the part of the M1 receptor that they are targeting. Studies using isolated cell membranes or recombinant proteins can identify antibodies that bind to any part of the protein. Because CHRM1 is an integral membrane protein with several transmembrane regions, only antibodies that bind to parts of the CHRM1 that are normally located on the outside of the membrane are likely to be of functional relevance in disease (as is seen in many other antibody-mediated neurological disorders). Patients could potentially have antibodies that bind to pieces of the protein that are normally present on the inside of the cell, but these would be unlikely to be pathogenic. The optimal assay to assess this would be a cell-based assay, using cells transfected with individual CHRMs, as the CHRM would then mimic the natural orientation of the receptor in the membrane. In addition, the antibody isotypes will also affect the functional capacity of the antibodies, as only certain isotypes will be able to exert functional effects such as reduction of receptor density or lysis of CHRM+ cells.
Some of the changes to CHRM expression and function observed in schizophrenia could plausibly be due to the effects of autoantibodies targeting these receptors. The lower levels of M1 receptors in brain tissue of MRDS cases, for example, could be due to antibody-mediated internalization of receptors, similar to that observed for myasthenia gravis, where CHRM expression at the neuromuscular junction is downregulated by internalization of antibody–receptor complexes into the neurons, where they can either be stored for later recycling back to the cell surface, or else be degraded (Tzartos et al., 1986). In addition, antibodies may cause receptor degradation, which would account for the loss of both membrane- and non-membrane-associated receptors in subjects with MRDS (Scarr et al., 2018a). It is of potential interest that the frequency of cases of MRDS (reported to be approximately 25%), is similar to the proportion of patients reported to carry elevated levels of anti-M1 receptor antibodies.
Anti-CHRM antibodies could also modulate cell signalling by acting as agonists or antagonists for CHRM-expressing cells. In support of this are the aforementioned studies showing agonist-like activities of anti-CHRM antibodies on neurons (Borda et al., 2002) and astrocytes (Borda et al., 2004). It might be thought that antagonist-like activity of antibodies would be more consistent with the muscarinic hypothesis of schizophrenia, in that antagonism of CHRM could lead to a decrease in muscarinic signalling. However, the overall effect could depend on the fine specificity of the antibodies (i.e. specificity for the activating M1/M3/M5 receptors vs specificity for the inhibitory M2/M4 receptors) and where the antibody acts. In addition, anti-CHRM antibodies could also initiate regulatory mechanisms leading to the upregulation or downregulation of the receptors on the neural cell surface. GPCR’s also feature self-regulatory functions to maintain homeostasis. For example, high levels of activation reduce the receptor’s ability to be stimulated in the future and vice versa (Gainetdinov et al., 2004). In addition, prolonged stimulation of a GPCR can also initiate downregulation via internalization, resulting in profound loss of receptor activity (Bohm et al., 1997). If the anti-CHRM antibodies are agonists with high avidity, they could induce an initial period of hypercholinergia, followed by hypocholinergia, once regulatory mechanisms are employed.
In addition, it is not yet known if anti-CHRM antibodies interact with orthosteric or allosteric binding sites. A selective PAM for the M1 receptor, BQCA, improved the efficacy of antipsychotic medications and reduced behavioural deficits seen in a rodent model of schizophrenia (Choy et al., 2016). Intriguingly, one of the binding sites for BQCA overlaps with the epitope recognized by anti-M1 receptor antibodies (Borda et al., 2002; Jones et al., 2014). Therefore, the possibility exists that M1 receptor antibodies could also block the allosteric binding site from a yet-to-be-identified naturally occurring ligand, or may themselves act as allosteric modulators. Immunocytochemistry using cultured cells expressing the M1 receptor could help to determine if the allosteric binding site is critical to the antibodies by comparing the relative antibody binding activity of cultured cells treated with and without BQCA.
Another mechanism that could potentially play a role in effects of anti-CHRM specific antibodies is antibody-mediated cell lysis. For example, the decreased density of M1 receptors in schizophrenia could be due to internalization of receptors, as discussed above, or to antibody-mediated killing of cells expressing these receptors. A 2% reduction in intracranial and total brain volume, mainly observed in grey matter structures, is consistently found in people with schizophrenia (Haijma et al., 2013). However, there do not appear to be significant decreases in neuronal cell numbers, suggesting that, if neuronal cell lysis is occurring, it is not widespread. An alternative hypothesis is that CHRM-expressing glial cells are decreased in number. There have been no studies comparing brain volume or cell (neuronal or glial) loss in those with and without anti-CHRM antibodies, to address this hypothesis. In MRDS, there is loss of CHRM1 positive neurons (Scarr et al., 2018a): antibody-mediated neuronal lysis could be one explanation for this observation; however, there is no research supporting this hypothesis.
Pathogenic antibodies directed towards the different subtypes of the CHRM could have differential effects based on their molecular activity and regional distribution. For instance, antibodies against the muscarinic inhibitory receptors (M2 and M4) could potentially contribute to a hypocholinergic phenotype, causing greater positive symptoms and cognitive dysfunction. Experiments using knockout mice have shown that the M3, M4 and to some extent M5 receptor subtypes are associated with changes in striatal dopamine release (Zhang et al., 2002), suggesting that antibodies directed against these subtypes could influence positive symptom severity in schizophrenia.
Future research
All of the studies on autoantibodies against CHRMs have been carried out in patients with long-term disease. Many antipsychotics are known to modulate immune function (Al-Amin et al., 2013) and to also regulate the density of neurotransmitter receptors (Han et al., 2008). Thus, the possibility exists that antibody levels found in those with chronic schizophrenia may be a consequence of long-term treatment with antipsychotic medications. In order to establish if this is the case, future studies should investigate CHRM antibodies in patients with first episode psychosis. Moreover, determining the presence of anti-CHRM antibodies at the onset of illness would assist in determining if they have pathogenic roles, or are reflective of the disease process. It is important that future studies use cell-based assays to allow a precise understanding of how these antibodies interact with muscarinic receptors. Cell-based assays would enable further research testing the ability of antibodies from patients with psychosis to modulate cell signalling or cause cell lysis in vitro, as well as determine the effects of antibody binding at the allosteric binding sites. In future trials, treatment with immunosuppressant could be considered for persons experiencing schizophrenia or psychosis associated with elevated levels of anti-CHRM1 antibodies. Before such trials could be initiated, it is essential that further research is conducted to determine the pathogenicity of anti-CHRM antibodies. As schizophrenia is a heterogeneous syndrome, it may be that sub-classifying patients with antibodies targeting CHRMs may help to inform the clinical presentation and treatment responses of a proportion of people living with schizophrenia.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Health and Medical Research Council (Grant 1101255). J.G.S. is supported by a National Health and Medical Research Council Practitioner Fellowship Grant (grant no.: APP1105807). A.R. and J.G.S. are employees of the Queensland Centre for Mental Health Research which receives core funding from Queensland Health. J.P.K. is supported by a University of Queensland Amplify Fellowship.