
Abstract
Background:
The blood–brain barrier acts as a highly regulated interface; its dysfunction may exacerbate, and perhaps initiate, neurological and neuropsychiatric disorders.
Methods:
In this narrative review, focussing on redox, inflammatory and mitochondrial pathways and their effects on the blood–brain barrier, a model is proposed detailing mechanisms which might explain how increases in blood–brain barrier permeability occur and can be maintained with increasing inflammatory and oxidative and nitrosative stress being the initial drivers.
Results:
Peripheral inflammation, which is causatively implicated in the pathogenesis of major psychiatric disorders, is associated with elevated peripheral pro-inflammatory cytokines, which in turn cause increased blood–brain barrier permeability. Reactive oxygen species, such as superoxide radicals and hydrogen peroxide, and reactive nitrogen species, such as nitric oxide and peroxynitrite, play essential roles in normal brain capillary endothelial cell functioning; however, chronically elevated oxidative and nitrosative stress can lead to mitochondrial dysfunction and damage to the blood–brain barrier. Activated microglia, redox control of which is mediated by nitric oxide synthases and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, secrete neurotoxic molecules such as reactive oxygen species, nitric oxide, prostaglandin, cyclooxygenase-2, quinolinic acid, several chemokines (including monocyte chemoattractant protein-1 [MCP-1], C-X-C motif chemokine ligand 1 [CXCL-1] and macrophage inflammatory protein 1α [MIP-1α]) and the pro-inflammatory cytokines interleukin-6, tumour necrosis factor-α and interleukin-1β, which can exert a detrimental effect on blood–brain barrier integrity and function. Similarly, reactive astrocytes produce neurotoxic molecules such as prostaglandin E2 and pro-inflammatory cytokines, which can cause a ‘leaky brain’.
Conclusion:
Chronic inflammatory and oxidative and nitrosative stress is associated with the development of a ‘leaky gut’. The following evidence-based approaches, which address the leaky gut and blood–brain barrier dysfunction, are suggested as potential therapeutic interventions for neurological and neuropsychiatric disorders: melatonin, statins, probiotics containing Bifidobacteria and Lactobacilli, N-acetylcysteine, and prebiotics containing fructo-oligosaccharides and galacto-oligosaccharides.
Introduction
The blood–brain barrier (BBB) is a highly regulated interface between the central nervous system (CNS) and the peripheral circulatory system. It has an indispensable role in maintaining homeostasis in the brain, and consequently, in brain functioning, influencing microglia activation as well as neuronal function and survival (Abbott et al., 2010; Hawkins and Davis, 2005; Jin et al., 2013). In particular, the BBB exerts tight regulation over the movement of ions, molecules and cells between the cells in the CNS and the blood (Daneman, 2012; Wong et al., 2013). It thus maintains the homeostasis of ions, hormones, neurotransmitters and the regulation of nutrients in the brain, while ensuring the segregation of neurotransmitters and other neuroactive molecules in the peripheral circulation and the CNS (Abbott et al., 2006; Luissint et al., 2012). The BBB also regulates the influx of immune cells and xenobiotics from the peripheral circulation into the brain and regulates the interstitial fluid (ISF) compartment (Abbott et al., 2010; Hawkins and Davis, 2005; Wong et al., 2013). Furthermore, the BBB plays an important role in the transport and metabolism of psychotropic agents used for the treatment of neurodegenerative and neuropsychiatric disorders (Abbott et al., 2010; Daneman, 2012; Wong et al., 2013). Unfortunately, many of the interactions that at physical and biochemical level maintain the integrity and function of the BBB, break down in the context of neuropsychiatric and neurological diseases. This paper aims to propose a model that can explain the mechanisms by which the increases in BBB permeability, seen in all neuropsychiatric (Najjar et al., 2013, 2017; Pollak et al., 2017) and neurological (Stanimirovic and Friedman, 2012; Takeshita and Ransohoff, 2015; Yamazaki and Kanekiyo, 2017) disorders, may occur.
BBB composition
All of the functions described above are enabled by the presence of highly specialized brain microvascular endothelial cells (BMECs) (Aird, 2007; Dejana, 2004). These possess highly organized tight junctions (TJs) and adherent junctions (AJs) as well as a range of specialized transporters, pumps and receptors. The TJs and AJs restrict the paracellular transport of polar substances, including hexose sugars, amino acids, nucleosides, monocarboxylic acids and vitamins (Grammas et al., 2011; Mokgokong et al., 2014). In addition, a plethora of specialized pumps and receptor transporters facilitate and regulate the entry, endocytosis and transendothelial transport of amino acids, nutrients and certain proteins such as insulin, leptin, transferrin and insulin-like growth factors, from the peripheral circulation into the brain (Abbott et al., 2010; Lajoie and Shusta, 2015; Meng and Takeichi, 2009; Ueno et al., 2010; Upadhyay, 2014).
AJs are formed by the haemophilic association between complementary members of the cadherin (calcium-dependent adhesion molecules) protein superfamily on the neighbouring membranes of BMECs. Catenins are found in complexes with cadherin molecules and include α-catenin and β-catenin subtypes. α-Catenin can bind to β-catenin. Cadherins are covalently linked to complementary members of the catenin superfamily in the cytoplasm. Catenins, in turn, are associated with several components of the cell cytoskeleton such as microtubules and actin filaments (reviewed by Harris, 2012; Hiroki, 2012; Meng and Takeichi, 2009).
TJs are primarily formed among membrane proteins called claudins (Jia et al., 2014), with occludins and other proteins playing a secondary role (Furuse and Tsukita, 2006; Hawkins and Davis, 2005). In the TJs, these proteins are anchored to the actin cytoskeleton via the zona occludin (ZO) adaptor proteins (ZO-1 and ZO-2) (Greene and Campbell, 2016; Hawkins and Davis, 2005). The claudin superfamily is composed of over 20 proteins, which are all indispensable players in TJ formation (Günzel and Yu, 2013). Of all the claudin superfamily, claudin-5, which has an important role in the regulation of paracellular ionic selectivity, is the most common isoform located in the BBB and the dominant player involved in the TJs (Hewitt et al., 2006; Jia et al., 2014), with claudin-s12 and -1 also playing a role, at least in some conditions (Abbott et al., 2006; Liu et al., 2012). It is important to note that the performance of TJs and AJs are structurally and functionally interdependent. For example, changes in the conformation and location of VE-cadherin result in upregulating transcription of the gene encoding claudin-5 (Dejana, 2004; Taddei et al., 2008).
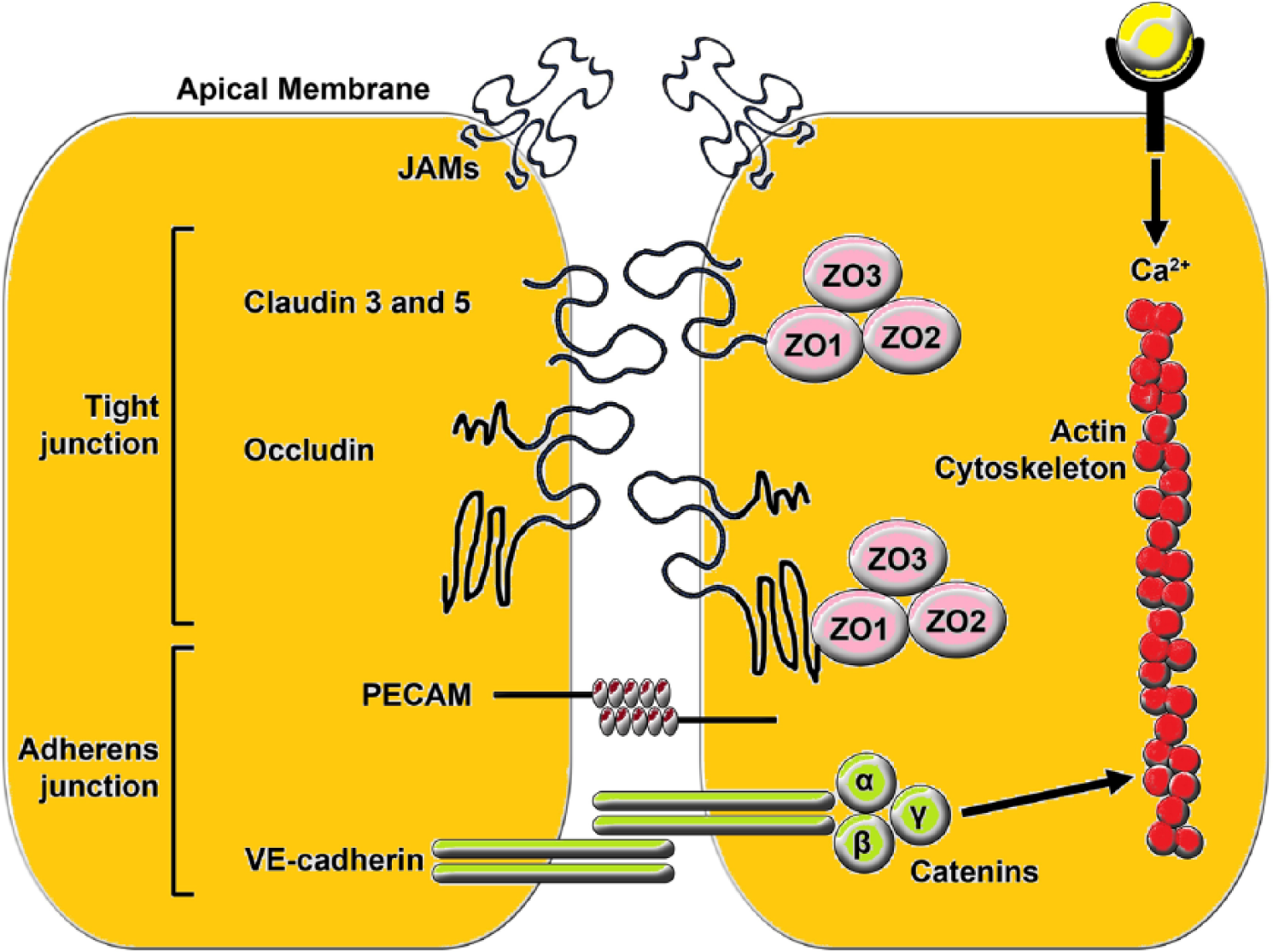
BMECs also possess a significantly increased density of mitochondria when compared with endothelial cells in the peripheral vasculature, which is likely reflective of the energy-dependent nature of their specialized transport roles (Lee and Pienaar, 2014; Nag, 2011). Structurally, BMECs are in intimate contact with pericytes and astrocytic end-feet, which ensheathes the brain vasculature, via the basal lamina, forming an additional continuous stratum, called the glia limitans, which separates blood vessels from the brain parenchyma. These astrocytes also enable contact between neurons, astrocytes, microglia, extracellular matrix components and myocytes (Hawkins and Davis, 2005; Stanimirovic and Friedman, 2012). The functional and signalling associations between these players enable the supply of blood to neurons to match changes in demand and form the neurovascular unit (NVU), which plays an indispensable role in maintaining the integrity and functional competency of the BBB (Hawkins and Davis, 2005; Najjar et al., 2013; Stanimirovic and Friedman, 2012). Figure 1 exhibits a diagammatic representation of the NVU. Figure 2 shows a representation of BMEC TJ.
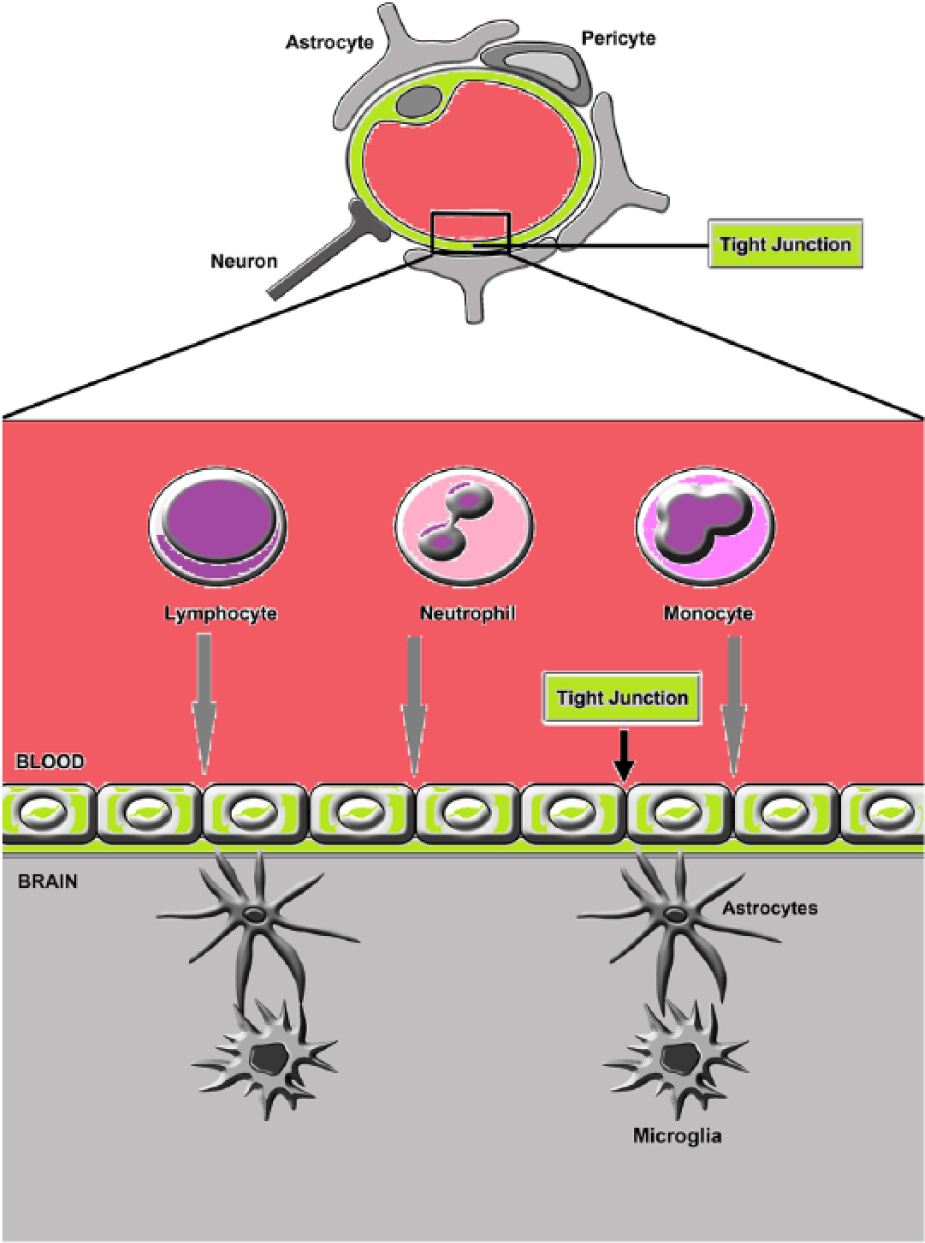
Diagrammatic representation of the NVU comprising the BMECs and the basal lamina, surrounded by pericytes astrocytic end-feet microglia and neurons.
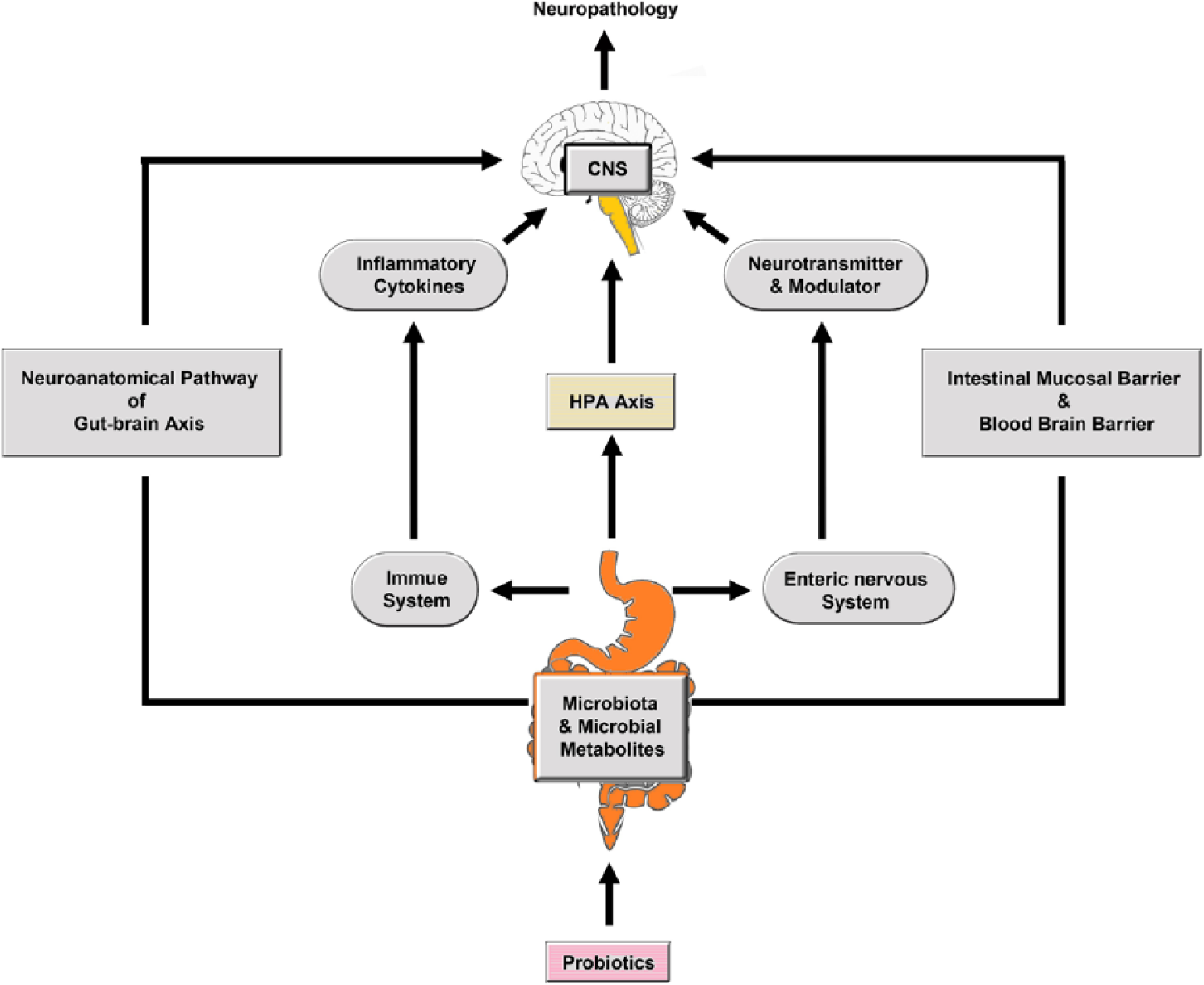
Schematic representation of the microbiota gut brain axis. There are five pathways involved in communicating between the microbiota and the brain. These are: the neuroanatomical pathway(represented by spinal afferent neurons and the vagus nerve), the neuroendocrine–HPA axis (facilitated by microbial production of hormones and neuropeptides), intestinal and systemic immune activation(characterized by LPS translocation and PIC production), altered permeability of the intestinal epithelium and blood–brain barrier (characterized by the production of SCFA and other metabolites) and, finally, the microbial production of neurotransmitters such as GABA and serotonin.
BBB and psychiatric disorders
Given the critical role of the BBB in neurophysiology, it is unsurprising that BBB dysfunction may play a role in neuropathophysiology including in the exacerbation and perhaps even the initiation of neurological illnesses (Stanimirovic and Friedman, 2012; Takeshita and Ransohoff, 2015; Yamazaki and Kanekiyo, 2017). Such neurological disorders include stroke (Sandoval and Witt, 2008; Ronaldson and Davis, 2012), Alzheimer’s disease (AD) (Banks, 2012; Zlokovic, 2011), multiple sclerosis (MS) (Miller, 2012; Zlokovic, 2008) and Parkinson’s disease (PD) (Bartels, 2011; Zlokovic, 2008). There are also accumulating data indicating that BBB disruption and/or dysfunction is involved in the pathogenesis and pathophysiology of psychiatric disorders such as schizophrenia (SZ) (Najjar et al., 2013, 2017; Pollak et al., 2017), major depressive disorder (MDD) (Najjar et al., 2013) and bipolar disorder (BD) (Patel and Frey, 2015).
Decreased BBB permeability and dysfunction of the NVU can be induced by peripheral inflammation in the guise of elevated pro-inflammatory cytokines (PICs) (Capaldo and Nusrat, 2009), peripheral and central oxidative stress via elevated reactive oxygen species (ROS) and reactive nitrogen species (RNS), (Najjar et al., 2013) neuroinflammation characterized by activated microglia and astrocytes (Tu et al., 2011), elevated levels of circulating lipopolysaccharide (LPS) (Yu et al., 2015), mitochondrial dysfunction (Doll et al., 2015), or even changes in the composition of the gut microbiota (Braniste et al., 2014). These elements also have an acknowledged causative role in the pathogenesis of neurodegenerative and neuroprogressive illnesses (Lucas et al., 2015; Morris and Berk, 2015; Morris et al., 2015a). Hence, the BBB disruption and/or dysfunction seen in persons suffering from such disorders may have multiple causes, which poses a significant challenge in the quest to develop an effective restorative therapeutic intervention.
Arguably, this quest has been hampered by the absence of an integrative model detailing the mechanisms whereby inflammation, oxidative stress, mitochondrial dysfunction, bacterial translocation, dysbiosis and neuroinflammation cooperate to cause, maintain and even accelerate increases in BBB permeability in neurodegenerative, neuroinflammatory and neuroprogressive diseases. There also seems to be a dearth of research aimed at uncovering which, if any of these elements might have primacy, and which might be classified as ‘downstream’. Understanding such mechanisms and their relative influence is an important step in the search for an effective therapeutic approach. In this context, it is interesting to note that inflammation and peripheral oxidative and nitrosative stress (I&ONS), which are invariant companions (Morris and Berk, 2015; Nafar et al., 2011; Vaziri, 2008), can induce BBB permeability (Morris et al., 2015a, 2015b) and intestinal barrier permeability (Al-Sadi et al., 2009; Banan et al., 2003; Lee, 2015; Tian et al., 2017) with the translocation of bacterial LPS and microbial metabolites into the peripheral circulation (Morris et al., 2016a, 2016b). In addition, peripheral I&ONS and LPS can also play a major role in the development of neuroinflammation (Morris et al., 2015a, 2015b). Accordingly, this paper aims to propose a model detailing mechanisms that explain how increases in BBB permeability might occur, and how they might be maintained, with elevated I&ONS being initial drivers of such permeability, both via direct effects on BBB endothelial cells, and indirectly by activating microglia and astrocytes in the brain. Other I&ONS-driven consequences include increased intestinal permeability with subsequent LPS translocation into the systemic circulation, and the likely development of dysbiosis. It is proposed that neuroinflammation, LPS translocation and dysbiosis conspire with chronically elevated I&ONS to produce the pathological consequences of BBB disruption. We will begin by examining the roles of I&ONS and then move on to examine the roles of activated microglia, LPS and then finally dysbiosis before suggesting therapeutic options based on underlying mechanisms driving BBB disruption.
Elevated inflammation and oxidative and nitrosative stress and increased BBB permeability
Peripheral inflammation and the role of PICs
Peripheral inflammation is causatively implicated in the pathogenesis of SZ, BD and MDD (Berk et al., 2013; Müller et al., 2015), which is relevant as acutely or chronically elevated levels of PICs in the periphery are a major cause of increased BBB permeability and the development of the neuroinflammatory cascade characteristic of neurodegenerative and putatively neuroprogressive disorders (Morris et al., 2015b). Some of the mechanisms underpinning this effect, such as inhibition of ZO-1 transcription and activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in BMECs, appear to be common to all cytokines described above (Capaldo and Nusrat, 2009). Other mechanisms appear to be specific to a particular cytokine and vary according to tissue type, its levels and time.
For example, one mechanism underpinning the detrimental effect of the PIC interferon (IFN)-γ on endothelial cell TJs involves its decreased levels provoking cellular mislocalisation of ZO-1 (Blom et al., 2015; Blum et al., 1997; Youakim and Ahdieh, 1999). However, IFN-γ also increases endocytosis of occluding claudin and junctional adhesion molecule-A (JAM-A) via increasing micropinocytosis into early recycling endosomes (Bruewer et al., 2005; Utech, 2005). This, in turn, leads to an efflux of these proteins away from the area of cellular contact leading to discontinuous or disorganized TJs, which can be seen by electron microscopy (EM) (Hall, 1998; Utech et al., 2006). This process involves a significant increase in actomyosin contractility secondary to IFN-γ-induced activation of the small GTPase RhoA and the subsequent upregulation of Rho-associated kinase (ROCK) (Utech et al., 2006). The latter enzyme, in turn, phosphorylates and activates myosin light chain kinase (MLCK), which engages in actin remodelling, leading to the increased actomyosin contractility described above (Hall, 1998).
RhoA activation and subsequent MLCK phosphorylation, in this case ultimately mediated by tumour necrosis factor (TNF)-α-induced activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), also appears to be a cause of increased paracellular permeability following acutely elevated levels of the latter transcription factor (Ma et al., 2004, 2005, Ye et al., 2006). However, evidence indicates that increased TJ permeability following exposure to chronically elevated levels of TNF-α appears to be mediated by mislocalisation of claudin-5 and JAM-1 into the cytoplasm and by decreased translation of occludin (McKenzie and Ridley, 2007). There is also accumulating evidence indicating that another mechanism underpinning TNF-α-induced increases in paracellular transport involves the downregulation of occludin levels (Lv et al., 2010; Wang et al., 2011).
Interleukin (IL)-1β increases BBB permeability via mechanisms that are common to other cytokines (reviewed by Alluri et al., 2016; Michael et al., 2016). For example, one mechanism underpinning IL-1β-induced BBB disruption also involves the upregulation of matrix metallopeptidase-9 (MMP-9) (Alluri et al., 2014). Prolonged elevation of IL-1β also increases paracellular transport in brain capillary endothelial cells (BCECs) by inducing β-catenin-mediated downregulation of claudin-3 (Haines et al., 2016) and via the upregulation of RhoA kinase-mediated MLCK phosphorylation hyperpermeability (Lapointe et al., 2010; Wu et al., 2016). IL-1β also increases transcellular transport by a mechanism involving upregulating phosphokinase C (PKC) isoforms, which stimulate endocytosis and membrane trafficking (Alvi et al., 2007). However, the mechanism of inducing BBB disruption by summoning neutrophils to the BBB would appear to be unique to IL-1β (Blamire et al., 2000; Joice et al., 2009; Scholz et al., 2007). Finally, IL-1β elevation can also increase BBB permeability indirectly by potentiating the adverse effects of TNF-α via engagement in paracrine signalling with this cytokine in a feed-forward loop (Didier et al., 2003).
Several transcellular BBB transport pathways can be adversely affected by systemic inflammation and oxidative stress. Examples include downregulation of transporters for organic anions (Wittmann et al., 2015a), monocarboxylates (Wittmann et al., 2015b), amino acids (Wittmann et al., 2015a), prostaglandin E2 (PGE2) (Akanuma et al., 2011) and leptin (Nonaka et al., 2004). Experimental evidence indicates that peripheral inflammation also influences the expression of the multi-functional efflux transporter P-glycoprotein (Pgp) encoded by the ABCB1 gene (Liu and Liu, 2014; Löscher and Potschka, 2005). The weight of evidence indicates that acute peripheral inflammation downregulates the expression of Pgp on the luminal and abluminal membranes of BMECs and on astrocytic end-feet (Fernandez et al., 2004; Hartz et al., 2006; Pardridge et al., 1997). Prolonged or chronic peripheral inflammation however appears to upregulate the expression of the transporter protein in the same regions (Liu and Liu, 2014). This may be of pathological importance as regional abnormalities in the expression of Pgp in the BBB appear to be a feature of neurological illnesses (Qosa et al., 2015). This also appears to be the case for SZ and MDD, with Pgp being upregulated in the temporal cortex, basal ganglia and hippocampus of the former illness and upregulated in the frontal and temporal regions in the latter (De Klerk et al., 2009, 2010). This upregulation could explain the development of drug resistance in MDD and SZ and the initial downregulation of Pgp in the early phase of inflammation could conceivably contribute to loss of CNS homeostasis and exaggerated neuropathology contributing to initial BBB disruption from the ‘inside’ (Müller, 2018; Sita et al., 2017). Preclinical data also suggest that peripheral inflammation may induce upregulation of receptors and cytosolic proteins responsible for the uptake of TNF-α (Osburg et al., 2002), monoamines (Wu et al., 2015) and insulin (Xaio et al., 2001), which may have additional detrimental effects on CNS homeostasis.
Chronically elevated oxidative and nitrosative stress and damage to the BBB
ROS, such as the superoxide radical (O2− or O2•−) and hydrogen peroxide (H2O2), and RNS, such as nitric oxide (NO or NO•) and peroxynitrite (ONOO−), play essential roles in cellular signalling in BCECs under physiological conditions (Morris et al., 2016c). NO derived from endothelial nitric oxide synthase (eNOS) also exerts protective effects on BMECs via a number of routes, including free radical scavenging (Förstermann, 2006; Najjar et al., 2013; Pan et al., 2005; Stuehr et al., 2004). However, higher levels of NO and ROS result in oxidative damage to lipids, proteins and deoxyribonucleic acid (DNA) resulting in escalating damage to endothelial cells and the BBB and a loss of the protective effects of NO derived from eNOS (Lucas et al., 2015; Morris and Maes, 2014). This process may be of relevance from a wider perspective so far as the pathogenesis of SZ and BD is concerned, as reduced levels and abnormalities in function or expression of eNOS appear to be related to several aspects of pathology in both illnesses (Burghardt et al., 2013; Reif et al., 2006). It is also noteworthy that several authors have adduced evidence associating endothelial dysfunction with the development of MDD (Lavoie et al., 2010). While there is no direct evidence that this phenomenon is caused by abnormal eNOS activity the high levels of oxidative stress seen in MDD patients makes this scenario quite likely.
Briefly, extra-endothelial NO production generated by the activity of neuronal nitric oxide synthase (nNOS) and inducible nitric oxide synthase (iNOS) in the extra-endothelial environment is increased in an environment of oxidative stress due to the positive modulation of the former by increased cellular calcium ion levels (Magenta et al., 2016) and the latter by increased levels of PICs and NF-κB signalling (Chuang et al., 2010; Galea et al., 1992). Elevated NO in combination with O2− in an environment of chronic ONS results in the synthesis of the powerful and excessively reactive oxidant peroxynitrite (NO• + O2•− → ONOO−), which can inflict massive damage on the vascular endothelium (Förstermann, 2006; Morris and Maes, 2014), and ultimately lead to frank disruption of BBB integrity (Ding et al., 2014; Stuehr et al., 2004). In addition, the activity of eNOS is compromised in conditions of elevated oxygen as a result of changes in levels of calcium ions, arginine and the essential cofactor tetrahydrobiopterin (BH4) (Burghardt et al., 2013; Mitchell et al., 2007; Montezano and Touyz, 2012). It should also be noted at this juncture that chronic peripheral inflammation can also impair endothelial eNOS function (Burghardt et al., 2013).
The mechanism underpinning reductions of BH4 levels involves ROS-induced oxidation of BH4 to dihydrobiopterin (BH2), thereby decreasing levels of this molecule in the endothelium (Najjar et al., 2013). The subsequent decrease in the BH4 to BH2 ratio inhibits the activity of eNOS while uncoupling arginine as its substrate and thus allowing engagement with molecular oxygen and increased production of O2 (Bouloumie et al., 1999; Moens and Kass, 2006; Najjar et al., 2013). As mentioned above, O2−, in turn, combines with NO to form ONOO−, thereby further increasing the oxidative conversion of BH4 to BH2, which further lowers eNOS activity in a positive feedback loop (Chen et al., 2010; Szabó et al., 2007).
Reduced eNOS activity can decrease endothelial NO levels resulting in reduced cerebral blood flow (Najjar et al., 2013; Toda and Okamura, 2012). The development of cerebral hypoperfusion may also be linked to impaired vasodilation that is mechanistically linked to reduced neurovascular eNOS-dependent NO biosynthesis (Li et al., 2016a; Liu et al., 2016; Najjar et al., 2013). Moreover, sustained cerebral hypoperfusion can further compromise endothelial mitochondrial oxidative function, increasing the formation of endothelial ROS (Aliev et al., 2010, 2014; Liu and Zhang, 2012), which in turn promotes eNOS uncoupling and lowers endothelial NO levels, thereby further reducing cerebral perfusion in a positive feedback loop (Antoniades, 2006; Chen et al., 2010; Lavoie et al., 2010).
Mitochondrial dysfunction, which appears to be an invariant feature of SZ, BD and MDD (reviewed by Morris and Berk, 2015) can also be induced by high levels of NO, peroxynitrite and ROS directly via oxidative damage to mitochondrial DNA, lipids and proteins, as well as by inhibiting enzymes of the electron transport chain (ETC) and the tricarboxylic acid (TCA) cycle (Morris et al., 2016c, 2017b). This is of importance as virtually every aspect of mitochondrial biology plays a significant role in maintaining the functions and integrity of endothelial cells in general and BMECs in particular (Doll et al., 2015).
In brief, mitochondria play an indispensable role in regulating endothelial cell signalling pathways by maintaining intracellular calcium ion homeostasis and varying the production of ROS (reviewed by Caja and Enriquez, 2017). There are also accumulating data suggesting that mitochondrial biogenesis dynamics, location and mitophagy also play a vital role in maintaining the optimal performance of these cells (reviewed by Kluge et al., 2013). Mitochondria also act as guardians of endothelial cells capable of sensing changes in the intracellular environment and protecting the cells against the ravages of oxidative stress by engaging a number of ‘defensive’ responses (Koziel and Jarmuszkiewicz, 2013). One such response is the upregulation of uncoupling protein production which results in reduced mitochondrial respiration, and impaired mitochondrial membrane potential resulting in diminished adenosine triphosphate (ATP) and ROS production (Koziel et al., 2015; Szewczyk et al., 2015). It is noteworthy that this defence involving upregulation of uncoupling proteins has been associated with a decrease in the permeability of the intestinal epithelial barrier (Zhang et al., 2012). However, upregulation of uncoupling proteins may be something of a double-edged sword as far as BMECs are concerned, as the function of membrane pumps and the integrity of TJs and AJs are dependent on an adequate supply of ATP (Bacallao et al., 1994; Mandel et al., 1993). Moreover, recent experimental data have demonstrated that profound mitochondrial dysfunction is associated with a dramatic increase in the permeability of the BBB, which is unsurprising given its energy-dependent nature (Bukeirat et al., 2015; Doll et al., 2015).
Indirect detrimental effects of chronic inflammatory & oxidative and nitrosative stress on the BBB
PICs can communicate inflammatory signals to the CNS via neural and humoral pathways to activate microglia and astrocytes, which can exert detrimental effects on the integrity of the BBB from the abluminal side via the production of ROS, RNS, PICs and a range of neurotoxic molecules (Morris and Berk, 2015). One humoral route involves direct access to the brain via circumventricular organs such as the subfornical organ which lack a functional BBB and the neurons of which are protected by being enwrapped by astrocyte-like stem cells (Morris et al., 2013; Perry and Holmes, 2014; reviewed by Miyata, 2015). Microglia proximate to circumventricular organs in general, and the subfornical organ in particular, are exquisitely sensitive to even slight increases in peripheral cytokine levels, and the intensity of their activation is excessive in relation to the strength of inflammatory stimuli and results in a range of cardiovascular and sympathetic responses (Furube et al., 2015; Wei et al., 2013). The weight of evidence suggests that such activation is initially defensive in nature and localized at low levels of peripheral inflammation, but becomes pathological and propagates throughout the CNS in wave-like patterns as levels of peripheral inflammation increase (Furube et al., 2018; Morris et al., 2013)
The other humoral pathways involve direct entry of PICs into the CNS via a saturable transport system in the BBB, or an indirect induction of cytokines and other inflammatory mediators such as prostaglandins and their subsequent release into the CNS parenchyma, or via provocation of an increase in BBB permeability (Morris et al., 2015b; Seruga et al., 2008). The neural route, on the other hand, involves direct stimulatory action of PICs on vagal afferent neurons (Goehler et al., 2000; Johnston and Webster, 2009). The stimulation of this nerve provides the main mechanism enabling the activation of microglia in the hippocampus following inflammatory insults such as the advent of an acute myocardial infarction (Francis et al., 2004a, 2004b). However, before moving on to consider the mechanisms underpinning BBB damage following the activation of microglia, it should be stressed that these glial cells display region-specific variation in the immune and bioenergetic pathways engaged during their ‘quiescent’ stage and following their activation (reviewed by Grabert et al., 2016 and Doorn et al., 2015). This is an important point as this state of affairs allows for considerable regional differences in the extent of BBB damage as a result of microglial activation driven by increased levels of peripheral I&ONS and could go some way to account for the regional variations seen in BBB disruption in diseases such as AD and PD (Gray and Woulfe, 2015; Zenaro et al., 2017), although disease-specific genetic and epigenetic factors are also involved (reviewed by Morris et al., 2017a). There is also evidence to suggest that the morphology, function and activation pattern of microglia in the brains of patients with SZ, BD and MDD display considerable regional variation (Jakobsson et al., 2015; Setiawan et al., 2015; Steiner et al., 2006, 2008; Watkins et al., 2014). Moreover, these parameters may also vary with illness state and clinical subtype, with the former allowing for considerable within-patient variation in the permeability of the BBB over time (Frick et al., 2013; Jakobsson et al., 2015; Laskaris et al., 2016).
Activated microglia and reactive astrogliosis as a cause of a ‘leaky brain’
Activated microglia secrete a range of neurotoxic molecules such as ROS, NO, PGE, cyclooxygenase (COX)-2, quinolinic acid, several chemokines such as monocyte chemoattractant protein-1 (MCP-1), C-X-C motif chemokine ligand 1 (CXCL-1) and macrophage inflammatory protein 1α (MIP-1α), and the PICs IL-6, TNF-α and IL-1β, which all exert a detrimental effect on the integrity and function of the BBB (Morris and Maes, 2014; Morris et al., 2013).
Redox control of activated microglia is mediated by NO synthases and NADPH oxidases (Rojo et al., 2014; Sumi et al., 2010). Microglial ROS induce BBB permeability via several different routes which include upregulation of the PI3K/Akt and c-Jun N-terminal kinase (JNK) signalling pathways, activation of MMP-9, MMP-3 and MMP-2 leading to cytoskeleton remodelling, and downregulation of TJ proteins claudins 5, 1 and 11 together with impaired transcription of ZO-1 and occludin (Asahi et al., 2001; Rosenberg et al., 2001; Schreibelt et al., 2007; Yamagata et al., 2004). Unsurprisingly, experimental evidence indicates that the peroxidation of membrane lipids in BCECs is another mechanism which underpins increases in BBB permeability mediated by ROS produced by activated microglia (Chodobski et al., 2011).
IL-1β, produced in copious amounts by activated microglia and astrocytes (Ravizza et al., 2008), mediates increases in BBB permeability directly by engagement with IL-1 receptor type 1 (R1), its signalling receptor; these receptors are expressed liberally on BMECs, perivascular astrocytes and microglia (Vezzani et al., 2008). Indirect increased BBB permeability is caused by IL-1β via upregulation of NO and several matrix metalloproteinases (MMPs) (Librizzi et al., 2012; Morin-Brureau et al., 2011), leading to redistribution of TJs and loss of ZO-1 with a resultant increase in BBB permeability (Obermeier et al., 2013). It is noteworthy that BBB damage ultimately originating from microglial derived IL-1 activates many downstream signalling pathways compromising many aspects of neuronal activity, particularly glutamatergic neurotransmission (Coulter and Eid, 2012; Kofuji and Newman, 2004). IL-1β activation also induces increased transcription of a wide range of adhesion molecules (e.g E-selectin, P-selectin, intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in BMECs. Upregulation of such molecules aids the adhesion of activated leukocytes at the luminal surface of these cells with the subsequent release damaging proteases and PICs ultimately allowing the entry of ions, proteins and other macromolecules from the periphery, thereby further compromising BBB integrity (Fabene et al., 2008; Kim et al., 2009).
MCP-1 plays an important role in the maintenance of BBB integrity in physiological conditions (Yao and Tsirka, 2011). However, evidence also suggests that this chemokine exerts a detrimental effect on BBB BCECs under neuroinflammatory conditions, provoking actin cytoskeleton remodelling and leading to the redistribution of occludin together with claudin-1, -5 and -11 (Dimitrijevic et al., 2006; Stamatovic et al., 2006).
Other molecular players released by activated microglia that play an important role in inducing BBB damage ‘from the inside’ include vascular endothelial growth factor (VEGF), IL-6, TNF-α, chemokine (C-C motif) ligand 2 (CCL-2) and prostaglandins. For example, IL-6 and CCL-2 play a major role in recruiting peripheral leukocytes which damage the BBB in the manner described above (Obermeier et al., 2013). VEGF, on the other hand, induces the downregulation of ZO-1 and promotes the angiogenesis and irregular proliferation of BCECs (Librizzi et al., 2012; Morin-Brureau et al., 2011).
Reactive astrogliosis and the development of a ‘leaky brain’
PICs and other neurotoxic molecules released from chronically activated microglia can induce activation, proliferation and a range of morphological and functional changes in astrocytes described as reactive astrogliosis. Perhaps unsurprisingly, these functional and morphological changes produce detrimental effects on BBB permeability and the integrity of the NVU (Cabezas et al., 2014; Chapouly et al., 2015). Reactive astrocytes produce a range of molecules capable of inducing BBB dysfunction or disruption, such as PGE2, IL-1β, IL-6 and TNF-α (Sofroniew, 2015). It is also worth noticing that chronic elevation of these cytokines leads to neurovascular uncoupling, or disruption of the relationship between local neural activity changes in cerebral blood flow. Neural activation normally is associated with neurotransmitter release as well as increased oxygen and ATP consumption. This releases vasoactive agents such as K+ and adenosine, which increase blood flow. Disruption of this process further increases BBB permeability and impairs the function of the NVU as a whole (Fujita et al., 2009; Giralt et al., 2010). Neurovascular uncoupling can also lead to increased levels of oxidative stress and a self-sustaining cascade of increased BBB permeability and frank disruption, mitochondrial dysfunction and oxidative stress, neuronal death and brain tissue atrophy (Lepore et al., 2008; Lewerenz et al., 2006).
Systemic LPS also induces reactive astrogliosis and may even induce apoptosis of these glial cells in certain circumstances (Biesmans et al., 2013; Cardoso et al., 2015). This leads to the disruption of the glia limitans and provides another mechanism underpinning BBB disruption (Asgari et al., 2015; Sofroniew, 2015). There are also data suggesting that LPS can induce disruptive structural changes in astrocytic end-feet, thus disrupting the architecture of the NVU per se (Fan et al., 2014). There is also accumulating evidence suggesting that systemically elevated LPS also provokes widespread changes in the transcription of astrocytic genes regulating cytotoxic and pro-inflammatory pathways, thereby increasing the production of PICs and other neurotoxins by these glial cells (reviewed by Zamanian et al., 2012). It is important to note that the development of reactive astrogliosis secondary to microglial activation, or indeed other inflammatory stimuli, is also associated with a dysregulated expression or a myriad of regulatory genes in these glial cells (Zamanian et al., 2012). This latter point is particularly pertinent from the perspective of SZ, BD and MDD, as discussed below.
Abnormal expression of several genes involved in the regulation of astrocyte function has been reported in SZ, which may adversely affect neurotransmission. Such genes also play a regulatory role in the function of the NVU, which appears to be impaired in SZ patients (Bernstein et al., 2015; Najjar et al., 2017). Similarly, decreased astrocyte density and function appear to be a feature of BD, which may well play a role in the unbalanced neurotransmission seen in this disorder. Notably, lithium and other medicines used to treat BD, such as carbamazepine and valproate, modify the expression of several astroglial genes leading to positive shift in astroglial signalling and CNS homeostasis. It is tempting to speculate that such changes could improve the structure and function of the NVU, although it must be stressed that, to date, there is no evidence to support this hypothesis (Peng et al., 2016).
Several research teams have reported changes in the levels of protein and messenger ribonucleic Acid (mRNA) for acknowledged astrocyte markers such as glial fibrillary acidic protein (GFAP), the water channel aquaporin-4 (AQP4), gap junction proteins (connexion-40 and connexion-43), the calcium-binding protein S100B and the excitatory amino acid transporters 1 and 2 in MDD patients. These observations are of relevance because of the indispensable role astrocyte function plays in maintaining the integrity and function of the NVU, which is known to be dysfunctional in MDD patients (Najjar et al., 2013; Rajkowska and Stockmeier, 2013).
The relationship between inflammatory and oxidative and nitrosative stress and ‘leaky gut’
Inflammatory and oxidative and nitrosative stress and the development of a ‘leaky gut’
Several authors have proposed the presence of dysbiosis and a dysfunctional microbiota–gut–brain axis in the pathogenesis and pathophysiology of SZ, BD and MDD (Kanji et al., 2018; Mangiola et al., 2016). Similarly, increased intestinal permeability and translocation of PS and other commensal antigens into the circulation have been demonstrated in each of these illnesses (Maes et al., 2012, 2013; Severance et al., 2013). The pathophysiological importance of this phenomenon in SZ and MDD is emphasized by data demonstrating a correlation between the levels of LPS in the systemic circulation and levels of peripheral inflammation (Maes et al., 2012, 2013; Severance et al., 2013). In this context, it is noteworthy that increased intestinal permeability can be initially caused by elevated I&ONS.
Chronically elevated I&ONS induces increases in intestinal permeability (Al-Sadi et al., 2009; Banan et al., 2003; Lee, 2015; Tian et al., 2017) leading to the translocation of LPS and other commensal antigens such as peptidoglycan and flagellin, which ultimately traverse from the gut lumen into the intestinal mucosa (Lucas et al., 2015; Morris et al., 2016b). This creates a vicious feed-forward loop which accelerates the pattern of localized and systemic inflammation via several different mechanisms (Delzenne and Cani, 2011; Zhang and Zhang, 2013).
LPS in the colon exacerbates intestinal inflammation and reduces the frequency of regulatory T cells (or Tregs), thereby increasing the expression of PICs (Im et al., 2012). Excessive levels of LPS in the colon also increase epithelial TJ permeability by increasing the secretion of IL-8 by intestinal epithelial cells (Angrisano et al., 2010). Translocated LPS also increases TJ permeability by inducing increased expression and changes in the location of toll-like receptor (TLR) 4 and cluster of differentiation (CD) 14 in enterocytes (Guo et al., 2013).
The development of gut inflammation has serious consequences including, but not limited to, the recruitment of macrophages into mucosal tissue from the peripheral circulation that also produce PICs that alter epithelial permeability. Chronic accumulation of LPS, and other inflammatory molecules such as peptidoglycan and flagellin, in the intestinal mucosa creates a self-amplifying feed-forward loop which exacerbates localized inflammation, further increasing intestinal permeability leading to the translocation of LPS and other commensal antigens into the blood stream (Delzenne and Cani, 2011; Zhang and Zhang, 2013). Prolonged translocation of LPS into the systemic circulation leads to the activation of TLR4 and TLR2 on antigen-presenting cells (APCs) and T lymphocytes and the development of chronic systemic inflammation and chronic immune activation, further increasing levels of systemic PICs with increased detrimental effects on BBB integrity and/or function (Morris et al., 2015a, 2015b). The potential pathogenic consequences of translocation of bacterial components into the systemic circulation is emphasized by data demonstrating that this phenomenon is a major contributor to the chronic systemic immune activation, inflammation and oxidative stress seen for example in HIV seropositive people (Brenchley and Douek, 2008; Shan and Siliciano, 2014). LPS translocation into the systemic circulation following the advent of dysbiosis and increased intestinal permeability is now considered to be a source of metabolic endotoxemia, increasingly appreciated as an important driver of pathogenesis and pathophysiology in type 2 diabetes mellitus, metabolic syndrome and MS (Cani et al., 2008, 2009; Puddu et al., 2014; Riccio and Rossano, 2015).
‘Leaky gut’ and the development of a ‘leaky brain’
Chronically elevated LPS exerts its adverse effects on the integrity and function of the BBB and the NVU via several different mechanisms. For example, LPS has been shown to induce BBB dysfunction via NADPH oxidase-derived ROS (Liu et al., 2012; Zhou et al., 2014). Other mechanisms include the upregulation of diffusible mediators such as NO and metalloproteinases (Qin et al., 2015; Wong et al., 2004). The presence of increased levels of this commensal antigen at the peripheral side of the brain endothelium also leads to the upregulation of COX and inflammatory intracellular signalling systems, which involves mitogen-activated protein (MAP) kinase signalling (Aid et al., 2010; Banks et al., 2015; Qin et al., 2015), MLC phosphorylation (possibly associated with MLCK transcription), and a change in and rearrangement of filamentous (F)-actin, which in turn may disrupt TJ assembly (He et al., 2011). There is also some evidence to suggest that LPS signalling induces mitochondrial dysfunction in highly energy-dependent brain endothelial cells (Doll et al., 2015).
Upregulation of MAP kinase signalling may also underpin LPS-induced damage to the brain endothelium, which involves disturbances to the integrity of endothelial cell membranes and mitochondrial damage which may ultimately result in frank apoptosis (Cardoso et al., 2012; Karahashi et al., 2009). This phenomenon may also stem from LPS-induced acceleration of glycocalyx degradation (Wiesinger et al., 2013). This glycoprotein lines the apical surface of the endothelium and there is accumulating evidence to indicate that it is an important player in maintaining barrier integrity and inhibiting paracellular transport (Woodcock and Woodcock, 2012; reviewed by Reitsma et al., 2007). The mechanisms underpinning LPS-induced degradation of glycocalyx remain to be fully delineated but there is evidence to suggest that this phenomenon is driven at least in part by secondary activation of TNF-α (Wiesinger et al., 2013), ROS (Moseley et al., 1997) and MMPs (Lipowsky, 2012).
LPS also increases the expression of calveolin-1 (Jiao et al., 2013; Martins, 2015), which is significant given the role of the latter in regulating endocytotic transport across BBB endothelial cells and BBB permeability (Gu et al., 2011). Briefly, the small number of endocytotic vesicles and caveolae in BBB endothelial cells relative to the peripheral vasculature, and subsequently a lower rate of transcytosis, is one of the mechanisms maintaining the relative impermeability of the brain endothelium (Nag, 2003). Caveolae are indispensable players in the endocytotic pathway and are largely composed of calveolin-1 (Feng et al., 2013; Simionescu et al., 2009). Crucially, LPS-mediated phosphorylation of calveolin-1 increases the number of caleveoli and endocytotic vesicles, resulting in increased transendothelial permeability of brain endothelial cells (Wang et al., 2015). This is an alternative mechanism underpinning BBB permeability, which appears to be of prime importance in an inflammatory environment (Cipolla et al., 2004; Lossinsky and Shivers, 2004). Furthermore, in such an environment, stimulation of vesicular processes, which promotes transcytotic leakage, may be the dominant form of initial BBB impairment (Banks et al., 2015) and precedes paracellular opening (Fleegal-DeMotta et al., 2009; Knowland et al., 2014). This is a complex argument and readers who are interested in the details are invited to consult the work of Jiao et al. (2011) and Krueger et al. (2013). Finally, evidence suggests that systemically elevated LPS can also contribute to the development of BBB permeability via the activation of microglia throughout the brain following initial activation of microglia adjacent to circumventricular organs such as the subfornical organ in much the same manner as peripheral PICs discussed above (Radler et al., 2014; reviewed by Furube et al., 2018).
The role of microbial metabolites in the genesis of a ‘leaky brain’
Increases in systemic and intestinal inflammation are associated with the development of dysbiosis (Rawls, 2007) and a concomitant decrease in bacterial genera such as Bacteroides Firmicutes, Ruminococcus and Faecalibacterium and Roseburia, which produce short-chain fatty acids (SCFAs) (Cantarel et al., 2015; Tremlett et al., 2016; Yamada et al., 2015; reviewed by Forbes et al., 2016). Notably, this pattern has been repeatedly demonstrated in MDD patients during periods of relapse and remission, and decreased levels of Faecalibacterium have also been reported in BD patients (Evans et al., 2017; Jiang et al., 2015; Zheng et al., 2016; reviewed by MacQueen et al., 2017). There is also evidence suggesting that the extent of such a decrease correlates with the severity of both depression and mania (reviewed by Evans et al., 2017). The situation in SZ, however, appears to be more complex as a recent study investigating the composition of the gut microbiota reported relatively increased levels of the Lactobacillaceae, Brucellaceae, Halothiobacillaceae and Micrococcineae, whereas levels of the Veillonellaceae family were decreased (Kelly et al., 2017). The authors of this study also reported that the increase in Lactobacillus group numbers correlated positively with the severity of psychotic symptoms displayed by patients in the study (Kelly et al., 2017). This may signal a departure from evidence of reduced SCFA producing bacteria in MDD and BD as members of the Lactobacillus group are held to encourage the production of SCFAs (LeBlanc et al., 2017). The patients in this study were, however, prescribed anti psychotics which are known to have their own independent effect on the composition of the microbiota, and even potentially upregulating levels of Lactobacillaceae (Bahr et al., 2015) rendering any study in this area carried out on patients who are not treatment-naïve very difficult to interpret (Kelly et al., 2017).
Reduced production of SCFAs is problematic on several counts. First, SCFAs of microbial origin play an indispensable role in the maintenance of intestinal barrier integrity via ligation of glucagon-like peptide (GLP)-43, which leads to the synthesis of GLP-1 and GLP-2 (Bischoff et al., 2014; Ferreira et al., 2014). Consequently, a relative paucity of SCFA producers can result in increased intestinal permeability by provoking detrimental changes in the distribution and localization of occludin and ZO-1, resulting in increased LPS translocation into the periphery and increased levels of inflammation (Cani et al., 2009; Morris et al., 2016a). Second, there is accumulating evidence that SCFA translocation into the peripheral circulation exerts a broadly anti-inflammatory effect by suppressing the activity of macrophages, dendritic cells (DCs) and T lymphocytes (Kim et al., 2014; Masui et al., 2013; reviewed by Sivaprakasam et al., 2016). Finally, the weight of evidence suggests that SCFAs play an indispensable role in the formation and maintenance of the BBB by modulating different pathways involved in the gut–brain axis (Braniste et al., 2014; Frohlich et al., 2016; Hoyles et al., 2017). The mechanisms underpinning these phenomena involve either direct interaction with the vagus nerve (Kimura et al., 2011) and the enteric nervous system (Obata and Pachnis, 2016), or engagement with BBB endothelial cells via translocation from the gut into the peripheral circulation (MacFabe, 2012; Morris et al., 2016a). Importantly, the evidence suggests that lowered levels of microbial SCFA production in the gut lumen and the peripheral circulation in a state of chronic intestinal and systemic inflammation compromise BBB function and/or integrity via a number of different routes (Braniste et al., 2014; Fessler et al., 2013; Frohlich et al., 2016; Hoyles et al., 2017). Figure 3 illustrates the microbiota gut brain axis.
Diagrammatic representation of BMEC tight junction. The main TJ proteins are the transmembrane occludins and claudins which form dimers with equivalent proteins expressed on adjacent endothelial cells. ZO-1 binds both claudins and occludins as well as JAMs while linking each to the cytoskeleton. TJs are further reinforced by binding between ZO-2 and ZO-3, the cingulin-linking protein and actin. AJs are composed of transmembrane cadherin proteins bound to α, β and γ catenins which in turn are linked to α-actinin, vinculin and, once again, the cytoskeleton protein actin.
There are accumulating data suggesting that one such route involves entry into BMECs via monocarboxylate receptors (MacFabe, 2012) and thereafter acting as histone deacetylase (HDAC) inhibitors. Rescuing such inhibition rescues histone acetylation and the acetylation of other proteins, thereby modulating the expression of genes and the function of a range of proteins playing an indispensable role in the performance of cellular signalling systems and organelles, and so modulating epigenetic processes (Morris et al., 2016a). There is accumulating evidence indicating that the HDAC activities of SCFAs are directly responsible for maintaining the permeability of the BBB in animal models of various neurological diseases by increasing the expression of occludin and ZO-1 (Li et al., 2016b; Park and Sohrabji, 2016). SCFA acting as HDACs may also exert protective effects on BBB integrity via more indirect routes such as increasing the resistance of brain endothelial cells to the corrosive effects of oxidative stress (Ferrante et al., 2003) and exerting a range of anti-inflammatory effects leading to reduced T cell, DC, neutrophil and macrophage activity, thereby reducing PIC and inflammatory chemokine activity, which may be of major importance in neuropsychiatric and neurological diseases (Aoyama et al., 2008; Smith, 2015; Tan et al., 2014).
Another potential mechanism underpinning the beneficial effects of SCFAs on BBB integrity involves engagement with aryl hydrocarbon (Arh) receptors, which are widely expressed on brain endothelial cells and throughout the CNS (Dauchy et al., 2008; Filbrandt et al., 2004; Jacob et al., 2011). Arh is a ligand-activated transcription factor that responds to planar aromatic hydrocarbons including cytochrome P450. Engagement and subsequent activation of Arh receptors on BBB endothelial cells results in a downregulation of connexin-43, which is an essential gap junction protein and, unsurprisingly, such downregulation is detrimental to BBB integrity (Andrysik et al., 2013; Kabatkova et al., 2015). From the perspective of this paper, it is noteworthy that this downregulation is mediated by activation of MAP kinase signalling and is potentiated by elevated TNF-α levels in an environment of chronic inflammation (Kabatkova et al., 2015). Connexin-43 is also an important player in maintaining immune quiescence within the CNS and reduced expression of this protein plays an independent role in the recruitment of immune cells from the periphery into the CNS by increasing the expression of chemokines and other chemoattractants, thereby further increasing BBB permeability and exacerbating any pre-existing neuroinflammation (Boulay et al., 2015; Lee et al., 2017). Neuroinflammation in the guise of activated microglia and astrocytes can be induced by elevated I&ONS and LPS in the periphery as previously discussed (Morris et al., 2013, 2015b). Importantly, the development and persistence of neuroinflammation is another cause of increased BBB permeability and/or disruption via several different mechanisms, which we will now consider.
Consequences of BBB disruption
BBB disruption allows the unregulated influx of peripheral blood mononuclear cells (PBMCs) of the innate and adaptive immune systems, including macrophages, DCs, B lymphocytes and T lymphocytes, into the CNS where they execute a range of pro-inflammatory effects, which can initiate and/or exacerbate neuroinflammation (Prinz and Priller, 2017). For example, the infiltration of CD4+ T subsets and their relative proportions have a crucial influence on the extent and polarization of microglial activation and consequent neuronal damage (Gonzalez et al., 2014; Lucin and Wyss-Coray, 2009). CD4+ T cells, such as Th1, Th17, gamma delta (γδ) T cells and granulocyte-macrophage colony-stimulating factor (GM-CSF) producing CD4+ T cells, play a major role in maintaining and exacerbating chronic neuroinflammation, thereby perpetuating neurodegenerative and neuroprogressive processes (Gonzalez and Pacheco, 2014). Th17 T cells would appear to be the most common subset entering the CNS is the earliest stages of increased BBB permeability, which are further stimulated by microglia, astrocytes and resident or infiltrated CNS macrophages, acting as APCs leading to further disruption of the BBB, both as a result of inflammatory mediators released by Th17 lymphocytes and increased microglial activation (Carson et al., 2006; Murphy et al., 2010; Iruretagoyena et al., 2006; Ye et al., 2006). This increase in BBB disruption, in turn, is thought to accelerate the entry of γδ T cells and Th1 lymphocytes and a self-perpetuating cascade of neuroinflammation and BBB disruption (Gonzalez et al., 2014).
There is also a growing awareness that cytotoxic CD8+ T cells play a major role both in the initial impairment of BBB integrity and the progress to BBB disruption via entry into the CNS and probably by stimulating the activation and/or proliferation of microglia and astrocytes, compromising the integrity of the NVU (Junker et al., 2007), as well as secreting the PIC IL-17 (Huber et al., 2013). This is a rapidly developing area of research, and readers interested in acquiring more details are invited to consult the work of (Pilli et al., 2017).
There is also accumulating evidence demonstrating that activated memory B cell infiltration into the CNS following BBB disruption and/or upregulation of integrins and selectins on the surface of BMECs is a significant contributor to increased neuroinflammation and neuropathology along several different dimensions
Unsurprisingly, myeloid DCs of peripheral origin also play a major role in maintaining or amplifying pathology in diseases associated with neuroinflammation and BBB disruption (Bossù et al., 2015). It is worthy of note that concentration of peripheral DCs in the CNS in physiological conditions is low, but is dramatically elevated in conditions of BBB disruption and a neuroinflammatory environment (Bulloch et al., 2008; Greter et al., 2005). Moreover, in many neuroinflammatory illnesses, this increase in DC levels in the CNS is accompanied by a corresponding fall in the numbers of DCs in the periphery, indicating that CNS DCs have their origin in the periphery (Ciaramella et al., 2013). DCs act as an additional source of neuropathology, in much the same way as effector B cell subsets, namely by further stimulation and polarization of T cells by acting as APCs and by secreting neurotoxic PICs (Ganguly et al., 2013; Ludewig et al., 2016).
Activated macrophages recruited into the CNS also induce or encourage the development of neuropathology and accelerated BBB disruption by acting as APCs and by the secretion of PICs. These monocyte derivatives also secrete a range of free radicals, MMPs and glutamate (Hendriks et al., 2005). However, the overall effects of macrophage infiltration into the CNS are somewhat unpredictable and depend on their polarization, often described as M1 (pro-inflammatory) or M2 (anti-inflammatory). Thus, influx of macrophages can have neurotoxic or neuroprotective consequences (reviewed by Vogel et al., 2014).
Initial recruitment and adhesion of activated neutrophils to the BBB in response to BMEC chemokine synthesis and secretion in inflammatory conditions plays a major role in the development of BBB damage. Such adhesion and subsequent transmigration of neutrophils across the BBB depend on upregulation of ICAM-1, integrins and P-selectin on BMECs (Bernardes-Silva et al., 2001; reviewed by Varatharaj and Galea, 2017). Once across the BBB, transmigration of neutrophils increases parenchymal tissue inflammation and promotes further BBB disruption via the secretion of inflammatory chemokines, cytokines, angiogenic factors, lytic enzymes and MMP-9. The actions of neutrophils stimulate increased recruitment of other PMBCs into the CNS, and a mutual interplay between CNS neutrophils, B cells and T cells ensures the long-term survival of each species (Ransohoff and Brown, 2012).
The effects of increased BBB permeability on peripheral immune and inflammatory pathways appear to be under-discussed, but there is increasing evidence supporting a pro-inflammatory effect (Bargerstock et al., 2014). For example, the entry of astrocyte-derived S100B into the peripheral circulation following BBB disruption may act as a damage-associated molecular pattern (DAMP) and activate TLRs expressed on APCs and elevate levels of peripheral inflammation (Bargerstock et al., 2014; Kanner et al., 2003). This molecule may also have the potential to act as a specific serum marker for BBB disruption, which is of interest given a virtual absence of reliable markers that are predictive of patients who are of increased risk of developing chronic neuropathology (Marchi et al., 2003).
Potential therapeutic approaches
Melatonin
Melatonin has demonstrable in vivo protective and/or restorative effects on the function and integrity of the BBB via several routes. Such routes include inhibition of TLR4/NF-κB signalling (Alluri et al., 2016; Hu et al., 2017), inhibition of MMP-9 (Alluri et al., 2016), inhibition of NADPH oxidase-2 (Jumnongprakhon et al., 2016), inhibition of AMP-activated protein kinase (AMPK) activity (Wang et al., 2017), inhibition of nucleotide-binding domain and leucine-rich repeat pyrin 3 domain (NLRP3) inflammasome assembly and/or function (Rahim et al., 2017) and variable levels of impact on silent information regulator 1 (SIRT1) (Zhao et al., 2015). There is also an accumulating body of evidence indicating that melatonin administration decreases intestinal permeability and exerts restorative effects on a ‘leaky gut’ (Eliasson, 2014; Mei et al., 2011).
Melatonin has a broadly anti-inflammatory effect in an environment of chronically elevated I&ONS and neuroinflammation (Carrillo-Vico et al., 2013) Importantly, from the perspective of the research questions addressed in this paper, there is a considerable body of data demonstrating that the therapeutic administration of melatonin attenuates inflammatory responses subsequent to the commensal LPS-mediated activation of TLR4 and consequent MyD88 (myeloid differentiation primary-response gene 88; an adaptor molecule) or TRIF (TIR-domain-containing adaptor protein inducing IFN-β) upregulation by LPS (Chuffa et al., 2015; Xia et al., 2012). In addition, melatonin therapy also appears to inhibit the activity of NF-κB by impairing the DNA binding capability of the molecule with a concomitant reduction in the activity of PICs and NLRP3 – both known to promote permeability of BBB and intestinal TJs as described above (Farez et al., 2015; Garcia et al., 2015; Tripathi and Jena, 2010). It is also noteworthy that in vivo evidence indicates that the dose of melatonin needed to achieve such effects is of the order of 50–100 mg daily (reviewed by Acuna Castroviejo et al., 2011; Cardinali et al., 2013), and conventionally prescribed doses of 1–5 mg daily would appear to produce no such benefits (Dowling et al., 2005; Medeiros et al., 2007).
Melatonin also acts as a potent scavenger of RNS, ROS, carbonate ions and a number of organic radical species (Morris and Maes, 2017). The antioxidant properties of melatonin also include the upregulation of catalase superoxide dismutase (SOD), glutathione reductase and glutathione peroxidase (Pandi-Perumal et al., 2013; Sharafati-Chaleshtori et al., 2017). Melatonin is also a positive modulator of mitochondrial performance by enhancing the activity of ETC enzyme complexes and by increasing mitochondrial ATP production (Cardinali et al., 2013; Ganie et al., 2016; Srinivasan et al., 2011).
Statins
There is a wealth of in vivo clinical evidence obtained from human studies of chronic illnesses demonstrating that statin therapy is associated with a reduction in plasma levels of C-reactive protein (CRP), IL-1, IL-6 and TNF-α (Albert et al., 2001; Ascer et al., 2004; Gilbert et al., 2017). Furthermore, several research teams have reported that statins reduce COX-2 and MMP-9 activity when used therapeutically in a range of inflammatory diseases (Massaro et al., 2009; Turner, 2005). It is also noteworthy that the clinical use of statins reduces NF-κB activation (Ortego et al., 1999) leads to the upregulation of thioredoxin, reduced glutathione (GSH) and other cellular antioxidant enzymes (Haendeler, 2004; Umeji et al., 2006) and improves the bioavailability of endothelial NO (Antoniades et al., 2011; McFarland et al., 2014). The mechanisms underpinning the anti-inflammatory effects of statins stem from their capacity to inhibit small GTPase prenylation with consequent downregulation of transcription factors such as activator protein 1 (AP-1) and NF-κB, and subsequent inhibition of PIC production (Greenwood et al., 2006; Smaldone et al., 2009). Additional anti-inflammatory actions of statins also stem from their capacity to downregulate the expression of suppressor of cytokine signaling 3 (SOCS3), CD40, IL-6, IL-8 and MCP-1 (Smaldone et al., 2009; Veillard et al., 2006).
Experimental evidence suggests that statins exert their antioxidant effects in the periphery and in the brains (Barone et al., 2011; Butterfield et al., 2012) of people with chronic disease via a number of different mechanisms. For example, some statins, most notably rosuvastatin, which is hydrophilic, inhibit the Rho kinase pathway, which is widely distributed in the CNS in general and BMECs in particular (Bond et al., 2015; Rawlings et al., 2009; Tonges et al., 2012). This is of interest given the role that activation of this enzyme plays in the development of TJ permeability and the exacerbation of the neuroinflammatory milieu (Tonges et al., 2012). Another route enabling statin-induced reductions in peripheral and central O&NS in patients with cardiovascular diseases involves the inhibition of Ras-related C3 botulinum toxin substrate 1 (RAC-1) (another small GTPase), which leads to reduced NADPH oxidase activity (Al-Shabrawey et al., 2008; Whaley-Connell et al., 2008; reviewed by Kwok et al., 2013). Yet another route involves upregulation of the Kelch-like ECH-associated protein 1 (Keap1) / nuclear factor erythroid 2-related factor 2 (Nrf2) pathway which is often described as acting as the master regulator of cellular antioxidant defences. This is likely the main pathway driving the upregulation of non-enzymatic and enzymatic antioxidants such as catalase and SOD as well as thioredoxin and GSH as discussed above (Gorrini et al., 2013; Habeos et al., 2008; Mrad et al., 2012). Several authors have also adduced evidence that administration of statins reduces the stability of membrane lipid rafts, thereby inhibiting the transduction of ROS-mediated signalling and downstream inflammatory pathways instigating cytokine and chemokine production (Hothersall et al., 2006; Wang, 2014). Statins also have the capacity positively to regulate mitochondrial biogenesis and oxidative phosphorylation via increasing the activity of AMPK, especially in an environment of chronic oxidative (Choi et al., 2008; Sun et al., 2006).
Several research teams have reported that the administration of various statins ameliorates the neurotoxic consequences of activated microglia and astrocytes, primarily by inhibiting the proliferation and phagocytic capacity of these glial cells and their production of PICs, ROS, RNS and other inflammatory mediators such as COX and PGE2, by inhibiting NF-κB and the small G-protein p21RAS (Kuipers et al., 2006; Li et al., 2009; Pahan et al., 1997).
There are also accumulating data suggesting that statin therapy increases the activity of eNOS in patients with atherosclerosis and a range of other cardiovascular diseases (Kilic et al., 2015; Ota et al., 2010). The weight of evidence suggests that the underlying mechanisms enabling this effect involve increasing the levels of eNOS phosphorylation, upregulating levels of BH4 and in some instances SIRT1 (Aoki et al., 2012; Hattori et al., 2003). It is also relevant that several research teams have reported that rosuvastatin and atorvastatin improve cerebral blood flow, once again via a mechanism involving the inhibition of Rho kinase (Rikitake et al., 2005; Su et al., 2014).
Several randomized, placebo-controlled, double-blind studies have shown that statins may decrease β-amyloid levels in the cerebrospinal fluid and improve cognitive function in AD patients in the early stages of their illness (Geifman et al., 2017; Shinohara et al., 2014; Simons et al., 2002). There is also evidence that chronic statin administration leads to decreased formation of β-amyloid in AD patients’ serum (Lee et al., 2013). However, in contrast, other authors reported that statin administration conferred no significant benefit on the progression of symptoms in early AD patients (Buxbaum, 2002). Several other authors have reported that statin therapy appears to have no effect on β- and tau-amyloid peptide (Höglund et al., 2005; Riekse et al., 2006; Sano et al., 2011). However, a number of studies have also produced evidence suggesting that prolonged statin use may be associated with a reduced risk of developing dementia (Sparks et al., 2005) and PD (Dufouil et al., 2005). Moreover, the authors of a recent systematic review concluded that statin use mitigated against cognitive decline in patients with mild cognitive impairment and early AD especially in patients carrying the APOE4 allele (Smith et al., 2017).
Probiotics and prebiotics
Rodent studies have demonstrated that probiotic treatments containing Lactobacillus, Escherichia coli and Bifidobacterium can reduce intestinal epithelial gut permeability by upregulating essential transmembrane TJ proteins (Patel et al., 2012; Qin et al., 2007; Zyrek et al., 2007). Examples of TJ proteins upregulated by specific strains of probiotics include occludin, claudin-2, cingulin and ZO-1 (Mennigen et al., 2009; Ulluwishewa et al., 2011; reviewed by Yan and Polk, 2011). Several probiotic species were also reported to improve epithelial function by increasing IgA and mucin protection, thus also improving the physical defences against the attack by commensal species on the gastrointestinal epithelium (Natividad et al., 2012; Tlaskalová-Hogenová et al., 2011). There is also evidence to suggest that at least some probiotic bacterial species protect the intestinal barrier by reducing the rate of epithelial cell apoptosis (Yan and Polk, 2011).
Some probiotic species exert anti-inflammatory and immunomodulatory effects (Konieczna et al., 2012). This is supported by a considerable body of research, most of which appears to have focused on preparations based on Bifidobacteria and Lactobacilli, and these have invariably demonstrated the capacity to modulate the systemic and intestinal immune responses and reduce the level of inflammation (Hardy et al., 2013; Kanauchi et al., 2013; Shokryazdan et al., 2017). It should be stressed, however, that such properties may extend to a wide array of other probiotics based on other commensal species (Konieczna et al., 2012).
This capacity also seems to extend to certain prebiotics, most notably formulations containing fructo-oligosaccharides and/or galacto-oligosaccharide (GOS) (Gori et al., 2011; Shokryazdan et al., 2017; reviewed by Pandey et al., 2015), which also appear to exert direct and beneficial effects on the brain (Savignac et al., 2016). This latter finding may well be of particular relevance in an environment of impaired BBB integrity in light of data produced by a more recent study which demonstrated a reduction in LPS-mediated increase in IL-1β levels in mice fed on a commercial non-digestible GOS preparation compared with a control sample (Savignac et al., 2016).
N-acetylcysteine
While N-acetylcysteine (NAC) does not appear normally to cross the BBB, it can protect from the effects of dysfunction of the latter. One method may entail hydrolysis of the NAC molecule to yield the amino acid cysteine, which can then be used to biosynthesize the tripeptide GSH, which consists of glutamic acid, cysteine and glycine, while another may involve the scavenging, by NAC, of radical species (Halliwell and Gutteridge, 2015).
Murine studies have shown that NAC has a neuroprotective action following traumatic brain injury; the mechanism appears to involve the inhibition of the normal increased cerebral levels of NF-κB, IL-1β, TNF-α and ICAM-1, that is, an inhibition of the cerebral inflammatory response (Chen et al., 2008). Wernicke’s encephalopathy, associated with a deficiency of thiamine, is associated with BBB dysfunction, which appears to be mediated by the caveolin-1 pathway being induced by oxidative stress; again, other murine experiments have pointed to a reduction in this dysfunction by NAC, which is associated with normalization of caveolin-1 levels (Beauchesne et al., 2010). In a further finding, which is of potential interest in the treatment of AD, it has been shown that NAC protects against inflammation-induced dysfunction of BBB low-density lipoprotein receptor-related (LRP)-1, which in turn prevents LPS-induced dysfunction there of transport of amyloid β-peptide (Erickson et al., 2012).
Wang et al. (2016) recently carried out a particularly informative series of murine experiments relating to the diabetic brain, affirming: that type 2 diabetes mellitus is associated with an increased blood level of the glycating molecule methylglyoxal and showing that brain ischaemia–reperfusion is stimulated by diabetes, with the size of cerebral infarcts correlating positively with the ratio of methylglyoxal to GSH in the brain, and negatively with the brain GSH concentration; that administration of NAC is associated with increased cerebral GSH levels and attenuation of ischaemia-reperfusion-induced cerebral infarction; and that the formation of protein carbonyls (promoted by oxidative stress) and methylglyoxal adducts is attenuated by NAC.
Acute hepatic failure is associated with hyperammonaemia, which in turn is associated with neuroinflammation and neuropsychiatric presentations, such as hepatic encephalopathy (Albrecht and Norenberg, 2006; Ott and Vilstrup, 2014). The ammonia crosses into BMECs, where it adversely affects the functioning and expression of breast cancer resistance protein (BCRP) by activation of ammonia–ROS–extracellular-regulated protein kinase-1/2 (ERK1/2) (Li et al., 2016c). Murine experimentation has recently shown that NAC (a ROS scavenger) restores the functioning and expression of BCRP (Li et al., 2016c).
Accumulating in vivo evidence indicates that NAC administration can exert positive effects on glutamatergic neurotransmission via NAC-induced stimulation of the cysteine-glutamate antiporter (system xc−) in glial cells (Durieux et al., 2015; Kupchik et al., 2012). Increased glutamate levels in the extra-synaptic space activates presynaptic mGluR2/3, which results in the inhibition of glutamate release into the synaptic cleft thereby mitigating the development of glutamate excitotoxicity (Dean et al., 2011; Kupchik et al., 2012). This property has attracted interest in NAC as a potential treatment for substance abuse disorder (SUD) as dysregulation of glutamatergic neurotransmission is considered to be a major element underpinning the development of craving, which appears to be a universal feature of addiction irrespective of the substance or behaviour involved (McClure et al., 2014).
Several large blinded RCTs have produced promising results in the area of cannabis and cocaine abuse, with a reduction in craving and drug intake in the former instance and a reduction in craving in the latter case which seems to have been limited to addicts already in a state of abstinence (Gray et al., 2012; LaRowe et al., 2013; Roten et al., 2013). These and other studies were examined in a recent meta-analysis which concluded that larger trials involving the use of NAC as an adjunctive treatment of SUD should be considered given the promising, though inconsistent, results achieved thus far (Duailibi et al., 2017). The use of NAC in the treatment of methamphetamine addiction appears to be worthy of special focus as there is reasonable evidence that its use can reduce craving in individuals addicted to the substance (Mousavi et al., 2015). Furthermore, additional compelling data from animal studies exists demonstrating that NAC at 10 mg/kg/day protects against and may even prevent, methamphetamine-induced destruction of dopaminergic neurons (Chandramani Shivalingappa et al., 2012; Fukami et al., 2004). Unsurprisingly, there has also been considerable interest in the use of NAC as an adjunctive therapy in a range of neurological and neuroprogressive disorders, for which, once again, the results of trials to date have been promising but not conclusive (Bavarsad Shahripour et al., 2014; Berk et al., 2014; Dean et al., 2011; Deepmala et al., 2015).
Summary and conclusion
The BBB acts as a highly regulated interface separating the CNS and the peripheral circulation. BMECs, with their attendant TJs and AJs, enable tight regulation to take place of the exchange of molecules between these two compartments. BBB dysfunction may exacerbate, and perhaps even initiate, neurological disorders such as stroke, AD, MS and PD. Similarly, it appears to be of importance in the pathogenesis and pathophysiology of psychiatric disorders such as SZ, MDD and BD.
Increased BBB permeability and NVU dysfunction can be induced by peripheral inflammation, which is associated with elevated PICs, ROS and RNS; neuroinflammation, associated with activated microglia and astrocytes and by increased LPS and mitochondrial dysfunction; and gut microbiota changes. In turn, all these factors are associated with the pathogenesis of neurodegenerative and neuroprogressive illnesses.
In this paper, the roles of I&ONS, LPS, dysbiosis and neuroinflammation in causing BBB dysfunction were detailed. In turn, it was shown that the consequences of such dysfunction include the unregulated influx of PBMCs into the CNS, causing pro-inflammatory actions. In light of the findings described in this paper, the following evidence-based therapeutic approaches for neurological and neuropsychiatric disorders were suggested: melatonin, statins, probiotics and prebiotics, and NAC.
In conclusion, multiple lines of evidence point to a concerning influence of both a leaky gut and dysfunctional BBB in the pathogenesis and pathophysiology of a wide range of neurological and psychiatric disorders. Interventions which address these two factors may prove therapeutic for the associated neurological and psychiatric disorders.