
Abstract
Objective:
The US Food and Drug Administration approval process for psychotropic drugs requires safety studies of carcinogenicity in animals. These studies are consistently conducted and provide a database for assessment of potential biological risk of carcinogenicity in humans. This report is a systematic review of that database for psychotropic drugs.
Method:
US Food and Drug Administration–approved registration data (‘package inserts’) were examined, where available, for all psychotropic drugs in the following classes: antidepressants, antipsychotics, benzodiazepines/sedative-hypnotics, amphetamines and anticonvulsants.
Results:
Overall, new generation (atypical) antipsychotics (90%, 9/10 agents) and anticonvulsants (85.7%, 6/7 agents) showed the highest evidence of carcinogenicity among psychotropic drugs classes assessed. Antidepressants (63.6%, 7/11) and benzodiazepines/sedative-hypnotics (70%, 7/10) were next, and stimulants (with the exception of methylphenidate) were last (25%, 1/4 agents). Overall, 71.4% of all drugs examined (30/42) showed evidence of carcinogenicity in 43.2% (38/88) of specific experimental studies.
Conclusions:
US Food and Drug Administration–based analyses demonstrate that almost all atypical antipsychotics and anticonvulsants are carcinogenic in animals, as are the majority of antidepressants and benzodiazepines and methylphenidate. These animal-based results are not sufficient to draw definitive conclusions in humans, but they provide data that could be acknowledged in the informed consent process of clinical treatment.
Introduction
The use of psychotropic medications has dramatically increased over the past two decades (Pincus et al., 1998). In the United States, in 2005, people treated with antidepressants were approximately 27 million, as compared to 13.3 million in 1996, corresponding to an overall increase of 74% (Olfson and Marcus, 2009). Recent available data (for calendar year 2009) indicate that total US retail prescriptions for psychotropic drugs exceeded US$380 million, corresponding to a net dollar cost of US$22 billion (Greenblatt et al., 2011). The largest share of psychotropic drug prescriptions occurs at the primary care level (Mojtabai and Olfson, 2008); usually for anxiety and depressive symptoms and their physical correlates (Linden et al., 1999). Given the extensive use of these agents, carcinogenic risk should be carefully weighed when prescribing long-term drug therapies. From a public health perspective, even a small increased risk factor, when applied to a large population, will produce many preventable cases of disease (Rose, 1981).
The US Food and Drug Administration (FDA) approval process for psychotropic drugs requires a series of safety studies. All drugs are examined with similar preclinical paradigms in these safety studies. Carcinogenicity is one of the assessed outcomes. In the published scientific literature, some concerns have been raised about the potential carcinogenicity of some psychotropic drugs (Brambilla and Martelli, 2009; Jacobs and Hatfield, 2013). However, clinical studies are heterogeneous and rarely control for potential confounding factors (Kripke et al., 2012; Pottegard et al., 2013). Preclinical studies are extensive and use different methodologies, making comparisons between studies difficult (Bucher et al., 1994; Watkins et al., 1992). Furthermore, the non-publication of negative studies might introduce bias when assessing the published scientific literature (Easterbrook et al., 1991). The FDA-based studies—where all agents are assessed in consistent ways—can provide a database that can be explored, thereby allowing for comparisons within and between drug classes.
We reviewed the FDA preclinical in vivo evidence to assess and compare carcinogenic risk among and between drug classes, with a focus on the most commonly used psychotropic drugs.
Methods
A systematic review of the FDA preclinical evidence was conducted to retrieve unpublished preclinical studies on carcinogenicity of the most common marketed psychotropic drugs. Data were obtained from the final package insert approved by the Center of Drug Evaluation and Research of the FDA and available on the official website of the FDA (www.fda.gov/AboutFDA/CentersOffices/OrganizationCharts/ucm347877.htm).
Data on carcinogenicity outcomes including tumor promoting activity and frequency of tumor incidence were retrieved from animal studies conducted in vivo. Descriptive analysis was carried out and findings pooled by drug class. The following drug categories were considered: antidepressants, antipsychotics, benzodiazepines/sedative-hypnotics, amphetamines and stimulants, lithium and anticonvulsants. For each drug, the number of studies reporting positive or negative risk of carcinogenicity was compiled (Tables 1–5).
Summary of the FDA preclinical studies on the carcinogenic risk of marketed antidepressants.
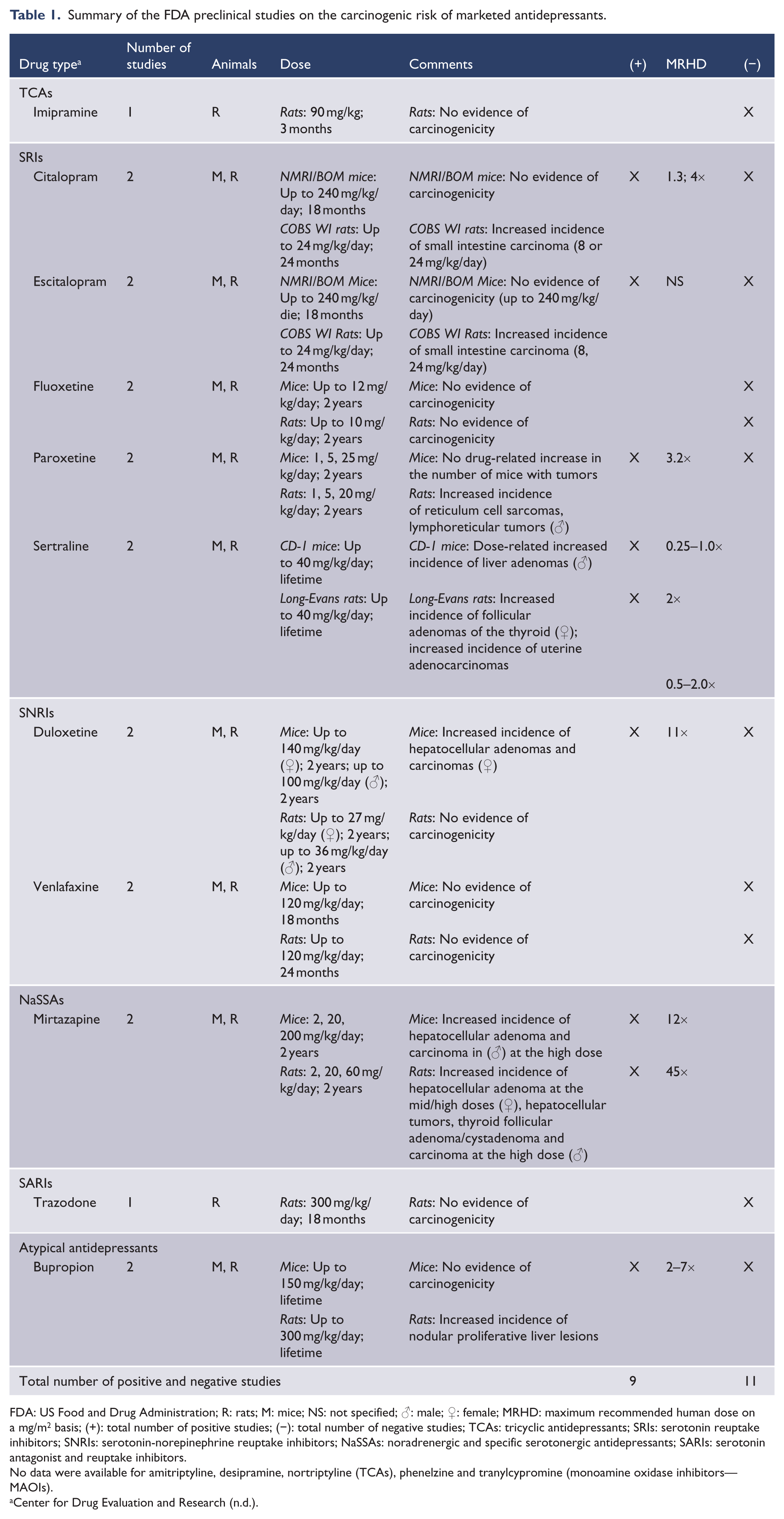
FDA: US Food and Drug Administration; R: rats; M: mice; NS: not specified; ♂: male; ♀: female; MRHD: maximum recommended human dose on a mg/m2 basis; (+): total number of positive studies; (−): total number of negative studies; TCAs: tricyclic antidepressants; SRIs: serotonin reuptake inhibitors; SNRIs: serotonin-norepinephrine reuptake inhibitors; NaSSAs: noradrenergic and specific serotonergic antidepressants; SARIs: serotonin antagonist and reuptake inhibitors.
No data were available for amitriptyline, desipramine, nortriptyline (TCAs), phenelzine and tranylcypromine (monoamine oxidase inhibitors—MAOIs).
Summary of the FDA preclinical studies on the carcinogenic risk of marketed antipsychotics.
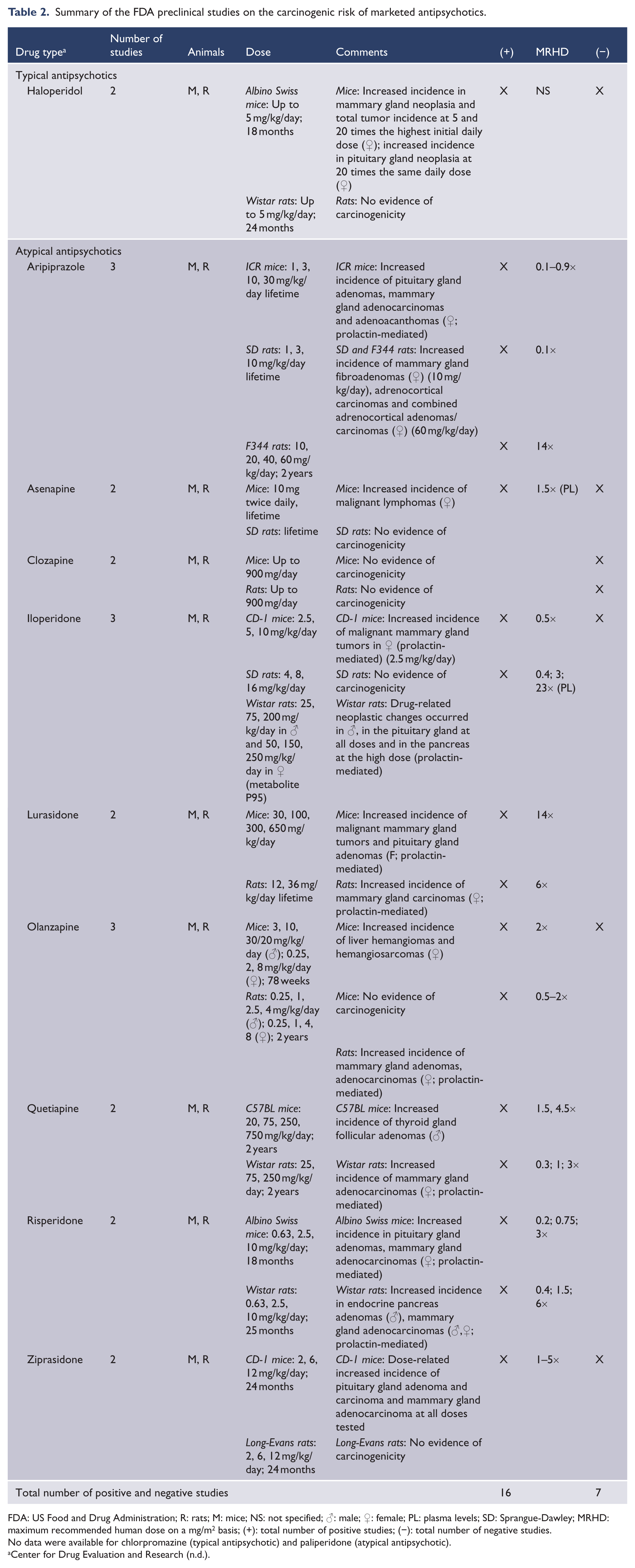
FDA: US Food and Drug Administration; R: rats; M: mice; NS: not specified; ♂: male; ♀: female; PL: plasma levels; SD: Sprangue-Dawley; MRHD: maximum recommended human dose on a mg/m2 basis; (+): total number of positive studies; (−): total number of negative studies.
No data were available for chlorpromazine (typical antipsychotic) and paliperidone (atypical antipsychotic).
Summary of the FDA preclinical studies on the carcinogenic risk of marketed benzodiazepines and sedative-hypnotics.
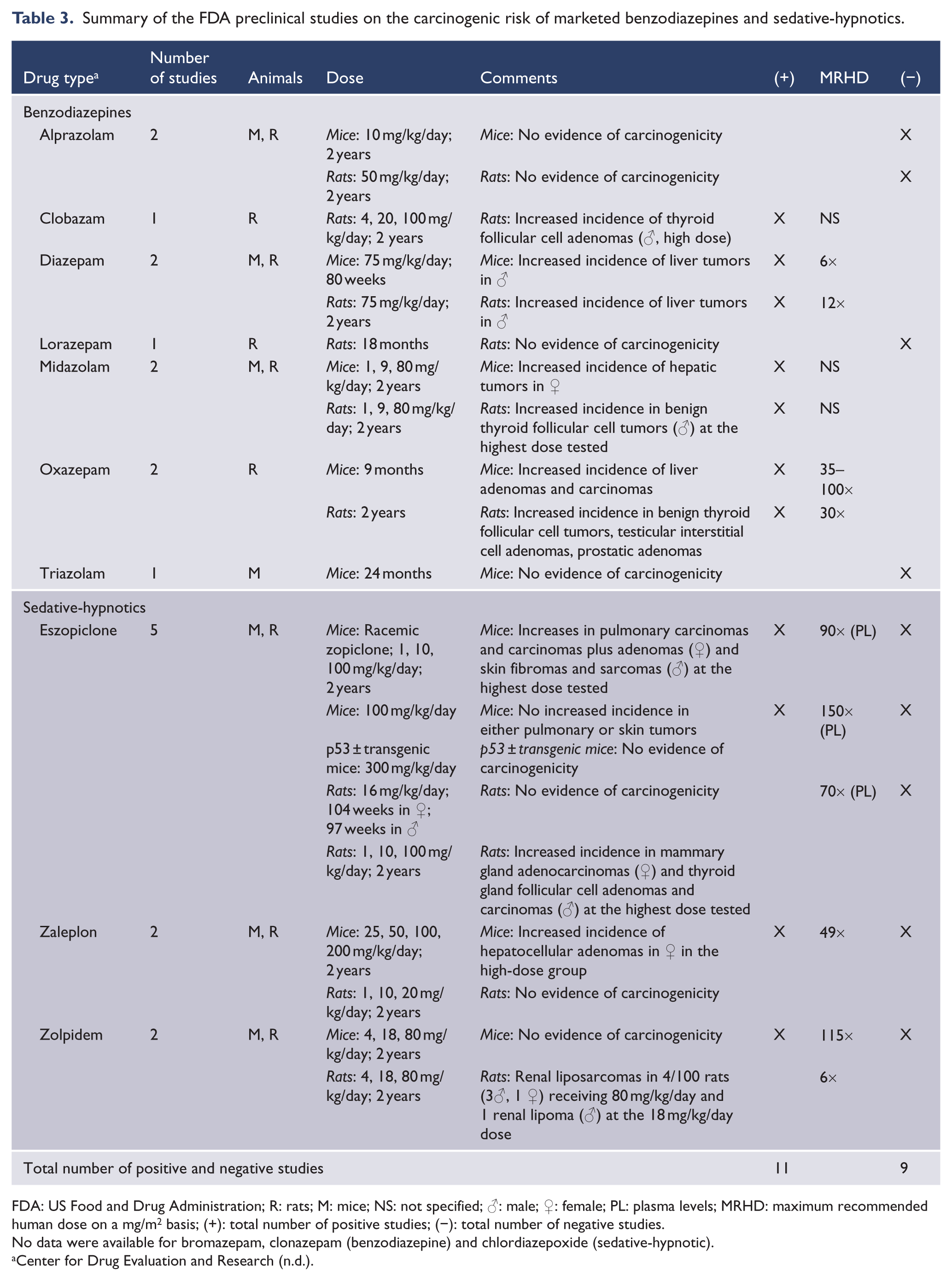
FDA: US Food and Drug Administration; R: rats; M: mice; NS: not specified; ♂: male; ♀: female; PL: plasma levels; MRHD: maximum recommended human dose on a mg/m2 basis; (+): total number of positive studies; (−): total number of negative studies.
No data were available for bromazepam, clonazepam (benzodiazepine) and chlordiazepoxide (sedative-hypnotic).
Summary of the FDA preclinical studies on the carcinogenic risk of marketed amphetamines and stimulants.
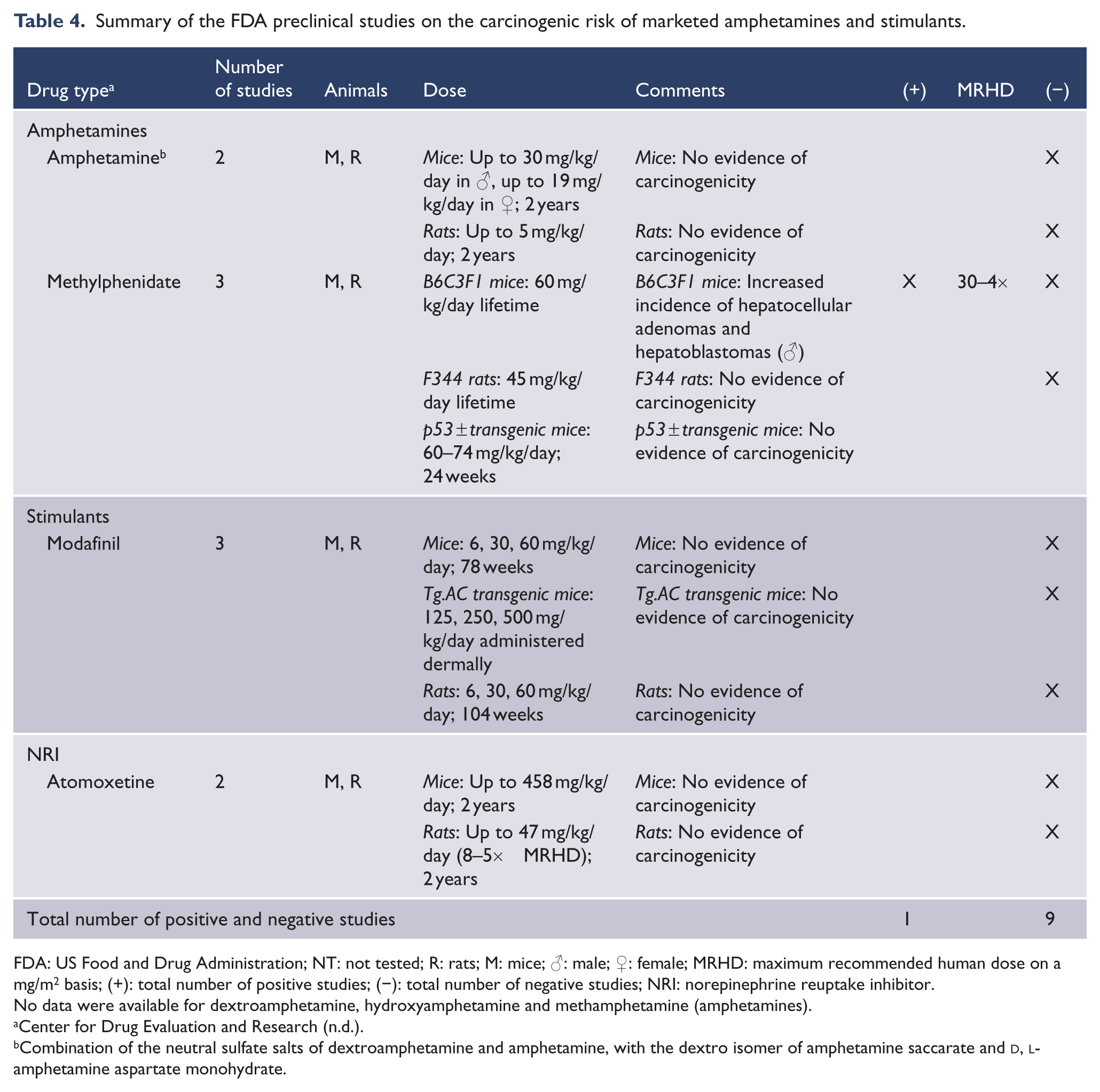
FDA: US Food and Drug Administration; NT: not tested; R: rats; M: mice; ♂: male; ♀: female; MRHD: maximum recommended human dose on a mg/m2 basis; (+): total number of positive studies; (−): total number of negative studies; NRI: norepinephrine reuptake inhibitor.
No data were available for dextroamphetamine, hydroxyamphetamine and methamphetamine (amphetamines).
Combination of the neutral sulfate salts of dextroamphetamine and amphetamine, with the dextro isomer of amphetamine saccarate and
Summary of the FDA preclinical studies on the carcinogenic risk of marketed anticonvulsants.
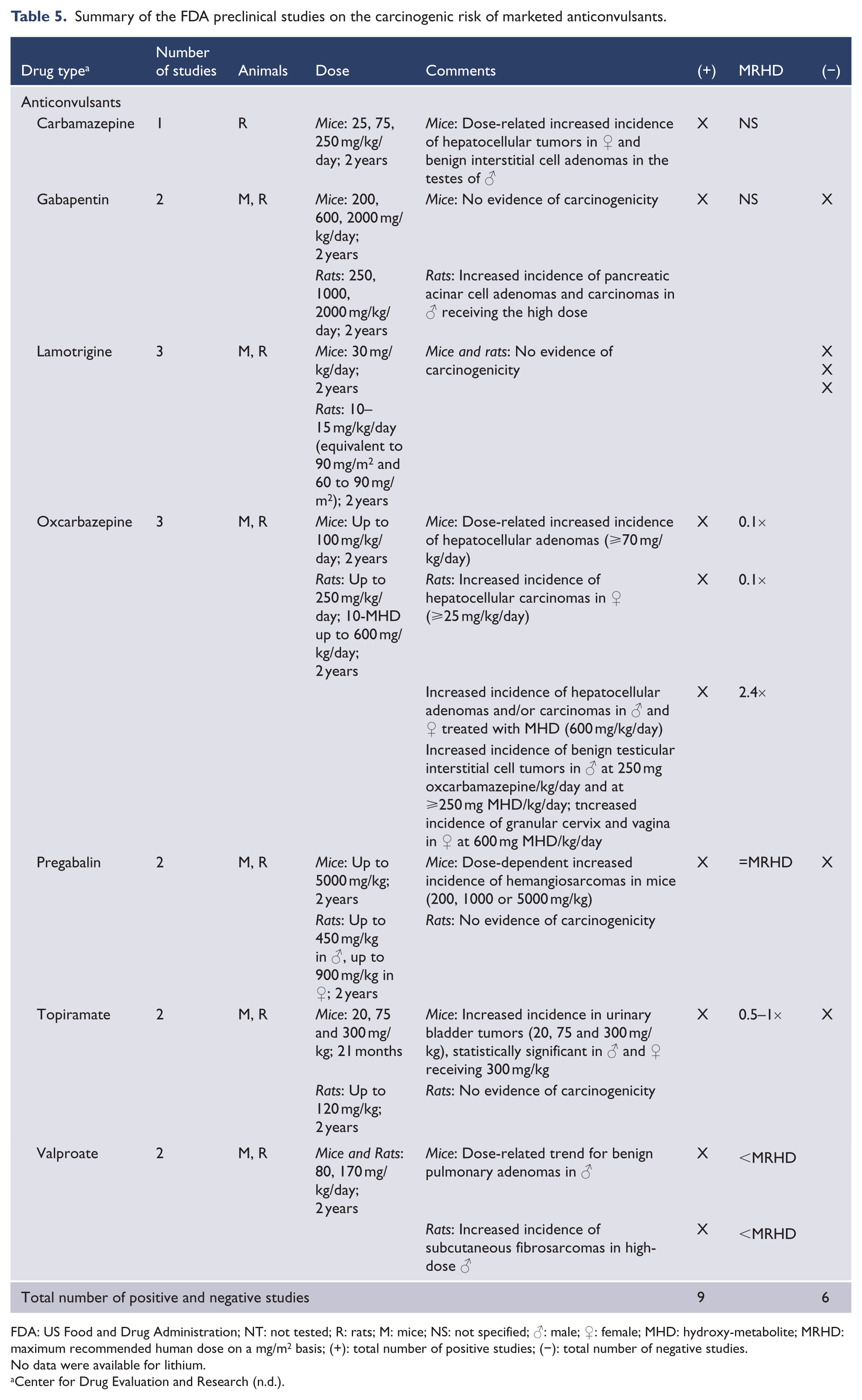
FDA: US Food and Drug Administration; NT: not tested; R: rats; M: mice; NS: not specified; ♂: male; ♀: female; MHD: hydroxy-metabolite; MRHD: maximum recommended human dose on a mg/m2 basis; (+): total number of positive studies; (−): total number of negative studies.
No data were available for lithium.
No data were available for some agents, mostly older drugs which entered the US market before current FDA efficacy and safety procedures were implemented in the 1960s and 1970s: amitriptyline, desipramine, nortriptyline (tricyclic antidepressants—TCAs); phenelzine, tranylcypromine (monoamine oxidase inhibitors—MAOIs); chlorpromazine (typical antipsychotic) and paliperidone (atypical antipsychotic); bromazepam, clonazepam (benzodiazepines) and chlordiazepoxide (sedative-hypnotic); dextroamphetamine, hydroxyamphetamine, methamphetamine (amphetamines); and lithium.
Results
If all examined psychotropic agents are combined, 71.4% (30/42) of agents had some preclinical evidence of carcinogenicity. Among antidepressants (Table 1), 63.6% (7/11) of examined agents were associated with carcinogenicity, with 45% (9/20) of all studies being positive for carcinogenicity. Specific agents associated with carcinogenicity were mirtazapine, sertraline, paroxetine, citalopram and escitalopram, duloxetine and bupropion. Agents unassociated with carcinogenicity were fluoxetine, venlafaxine, trazodone and imipramine. Among antipsychotics (Table 2), 90% (9/10) of examined agents were associated with carcinogenicity, with 69.6% (16/23) of all studies being positive for carcinogenicity. All agents were associated with carcinogenicity except clozapine.
Among benzodiazepines and sedative-hypnotics (Table 3), 70% (7/10) of examined agents were associated with carcinogenicity, with 55% (11/20) of all studies being positive for carcinogenicity. Specific agents associated with carcinogenicity were clonazepam, zolpidem, zaleplon, diazepam, eszopiclone, oxazepam and midazolam. Agents unassociated with carcinogenicity were lorazepam, alprazolam and triazolam.
Among amphetamines and stimulants (Table 4), 25% (1/4) of examined agents were associated with carcinogenicity, with 10% (1/10) of all studies being positive for carcinogenicity. Only methylphenidate was associated with carcinogenicity, in one-third studies of that agent. Amphetamine salts, modafinil and atomoxetine were unassociated with carcinogenicity.
Among anticonvulsants (Table 5), 85.7% (6/7) of examined agents were associated with carcinogenicity, with 64.3% (9/14) of all studies being positive for carcinogenicity. The only agent not associated with carcinogenicity was lamotrigine. Specific agents associated with carcinogenicity were valproate, carbamazepine, gabapentin, pregabalin, oxcarbazepine and topiramate. No data were available for lithium in the FDA-based safety information.
Discussion
Among psychotropic drugs classes, new generation (atypical) antipsychotics and anticonvulsants showed the highest evidence of carcinogenicity, with almost all agents (with the exception of clozapine and lamotrigine) being carcinogenic in FDA-based preclinical studies. A majority of antidepressants (63.6%), benzodiazepines and sedative-hypnotics (70%) were also found to be carcinogenic. Among the amphetamines/stimulant drug class, only methylphenidate had some evidence of carcinogenicity. If all examined psychotropic agents are combined, about 71% of agents had some preclinical evidence of carcinogenicity.
These results need to be interpreted in the context of the standards of assessment of potential human carcinogenicity with animal-based studies, as well as available evidence in human studies. Each aspect, standards of study of animal carcinogenicity and human studies themselves, will be considered in turn.
Animal species and strains
FDA preclinical in vivo studies to evaluate the carcinogenic potential of drugs are performed in mice and rats, in both sexes, and often extended over the average lifetime of the species (18 to 24 months for mice; 24 to 30 months for rats) (Hayes et al, 2011). To ensure that 30 rats per dose survive the 2-year study, 60 rats per group per sex are often started in the study. It is important to differentiate between different strains of rats and mice. As reported by recent National Toxicology Program (NTP) studies, Fisher 344 (F344) rats and B6C3F1 mice present high incidence of testicular tumors and leukemias, and liver tumors respectively (Casarett and Klaassen, 2008). Important choices also include the number of drug doses, and dose levels, and details of the required histopathology. Both gross and microscopic pathological examinations are performed not only on animals that survive the drug exposure but also on those that die prematurely.
Drug doses
To test the carcinogenic potential of drugs high drug doses are necessary. This is because of statistical and experimental design limitations of chronic bioassays. Identifying with statistical confidence a 0.5% incidence of cancer (over 1 million additional cancer deaths per year in the United States) in a group of experimental animals would require a minimum of 1000 test animals, this assuming no tumors were present in the absence of exposure. To test the potential carcinogenicity of a drug at the doses usually encountered by people would require using an impractically large number of animals. One alternative to this is to assume that there is a relationship between the administered dose and the carcinogenic response and administer to animals doses of drugs that are high enough to produce a measurable tumor response in a reasonable size test group (40 to 50 animals per dose) (Casarett and Klaassen, 2008).
Methodological rigor
FDA-required preclinical in vivo studies follow the Organisation for Economic Co-operation and Development (OECD) Guidances for Testing of Chemicals and the Good Laboratory Practice (GLP) for Nonclinical Laboratory Studies Guidelines (http://www.fda.gov/Drugs/DrugSafety/default.htm). This guarantees their methodological rigor.The OECD Guidances for the testing of chemicals are a collection of the most relevant internationally agreed testing methods used by governments, industry and independent laboratories to assess chemical products’ safety. They are primarily applied in regulatory safety testing and subsequent chemical notification and registration. OECD test guidelines should not be confused with data requirements, which are the prerogative of national authorities.The GLP for Nonclinical Laboratory Studies regulations set the minimum basic requirements for study conduct, personnel, facilities, equipment, written protocols, operating procedures, and study reports. Facilities conducting studies in accordance with the GLP regulations are required to have a Quality Assurance (QA) Unit to monitor each study so as to guarantee the quality and integrity of safety data in support of FDA-regulated products. The final study report should include a signed statement from the QA Unit with the dates the study was inspected and findings reported.
In addition, since 1990, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) bring together the regulatory authorities and pharmaceutical industry of Europe, Japan and the US to discuss scientific and technical aspects of drug registration and to ensure that safe, effective, and high quality medicines are developed and registered in the most resource-efficient manner.
Relevance to humans
Transposing animal studies’ findings to humans remains controversial (Hackam and Redelmeier, 2006). Rats and mice give concordant positive or negative results in only 70% of preclinical studies; it is therefore unlikely that the concordance between studies conducted on, respectively, rodents and humans would be higher (Lave et al., 1988). In addition, the rodents that are commonly used to study experimental tumors’ growth have relevant genetic, molecular, immunologic and cellular differences as compared to humans. Further, despite the recent development of transgenic mouse models to better identify carcinogens and mechanisms of action (Bucher, 1998), animal models are unable to entirely replicate the complex physiological changes that occur in humans’ physiopathology (Mak et al., 2014).
However, preclinical studies in animal are still a key component of the hazard identification process. All human carcinogens that have been adequately tested in animals produce positive results in at least one animal model. Thus, “although this association cannot establish that all agents and mixtures that cause cancer in experimental animals also cause cancer in humans, nevertheless, in the absence of adequate data on humans, it is biologically plausible and prudent to regard agents and mixtures for which there is sufficient evidence of carcinogenicity in experimental animals as if they presented a carcinogenic risk to humans”. (International Agency for Research on Cancer (IARC) and World Health Organization, 2000).
Available human data
These results must be interpreted in the context of the published scientific literature. Findings from published preclinical animal studies are heterogeneous, with some studies showing reduced cancer risk for some drug classes, such as antidepressants, based on proven beneficial immunological effects (e.g. enhanced natural killer cell activity) (Chou et al., 2007; Frick et al., 2008). Other studies, as in the FDA-based studies, show direct carcinogenic activity in animals exposed to some antidepressants (Brandes et al., 1992; Iishi et al., 1993).
Findings from clinical studies also are very heterogeneous. The only meta-analysis on the topic assessed the association between use of antidepressants and risk of breast and ovarian cancer, and reported a relative risk of 1.11 (95% confidence interval [CI]: [1.03, 1.20]). As the authors claimed, this small increase in risk might be important at the population level. In addition, they could not rule out the risk of publication bias (Cosgrove et al., 2011). Nevertheless, an 11% increase in risk can easily be accounted for potential confounding factors that exist in observational practice, such as smoking, diet and other environmental risk factors, none of which was controlled in the meta-analysis. Specific clinical studies, some of which are positive (Cotterchio et al., 2000; Dalton et al., 2000) and others negative (Coogan et al., 2005; Haukka et al., 2010), are hampered by similar methodological limitations.
The FDA-based analysis presented here has the advantage of being methodologically consistent for all agents and unaffected by publication bias. Furthermore, by being limited to preclinical animal settings, where other environmental risk factors can be controlled or minimized, it is less liable to confounding bias. While the relevance of these animal findings for human studies remains to be established, these basic science findings should not be ignored. The case of hormone replacement therapy (HRT) suggests that a widely used drug class may cause carcinogenicity, eventually proven by very large human randomized studies, which went previously undetected for decades despite widespread clinical usage and large observational clinical studies (Rossouw et al., 2002).
In sum, this FDA-based analysis, limited to animal studies, is not sufficient to draw definitive conclusions in humans, but it provides relevant animal-based data that could be acknowledged in the informed consent process of clinical treatment.
Footnotes
Declaration of interest
Dr Amerio, Dr Gálvez, Dr Odone and Mrs Dalley have no potential conflicts of interest to declare. Dr Ghaemi has provided research consulting to Sunovion and Pfizer and has obtained a research grant from Takeda Pharmaceuticals.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.