
Abstract
Objective:
The primary focus of this review is to provide an overview of the role of inflammation in the development of depression. The article will describe how inflammatory cytokines contribute to depression via action on three major pathways in the brain: the neuroendocrine; neurotransmitter depletion; and neuroprogression pathways.
Methods:
An online literature search was carried out in July 2012. Original articles and reviews were selected if they discussed the role of inflammation on the development of depression.
Results:
There is a large body of current research on the role of inflammatory cytokines on the development of depression. Cytokines have been found to interact with different pathways in the brain, and may contribute to the development of depression. Cytokines cause hypercortisolaemia by dysregulation of the hypothalamic–pituitary–adrenal axis directly by activating it and indirectly by modifying glucocorticoid receptor sensitivity to cortisol leading to cortisol hypersecretion. Cytokines deplete central synaptic serotonin levels by reducing its synthesis and increasing its reuptake. They may also deplete neurotrophic factors which are believed to play a neuroprotective role against depression. Cytokines activate cellular cascades that cause excitotoxicity and apoptosis and inhibit neurogenesis in the hippocampus.
Conclusion:
There is a growing body of correlative studies that suggest inflammatory cytokines may be a central factor that can affect multiple neuronal pathways and have an additive effect on the development of depression. However, the fact that not all people with inflammatory conditions suffer from depression suggests that depression is not purely a result of elevated inflammatory cytokines. Depression may be a result of a complex pathology that remains an area of growing interest and importance.
Introduction
Major depressive disorder (MDD) is a heterogeneous psychiatric illness, with a lifetime prevalence of approximately 12% and 20% in men and women respectively (Kessler et al., 2003). It is a long-term, relapsing condition in which 75% of patients experience more than one episode during their lifetime (Palazidou, 2012). It is currently the third leading global burden of disease and expected to become the second leading cause in the next decade (Mathers and Loncar, 2006).
There have been many theories regarding the aetiopathophysiology of depression, the most well-known being the “monoamine hypothesis” (Raedler, 2011). However, this theory alone, fails to explain the heterogeneity inherent in MDD amongst different patients and its variable response to treatment, thus highlighting our lack of understanding of this complex disorder. Over 30% of patients with depression fail to respond to conventional antidepressants (Maes et al., 2009; Miller et al., 2009a). Some theories which try to explain its pathophysiology include: the monoamine hypothesis, based on a neurotransmitter deficiency of monoamines, namely serotonin and noradrenalin; the hypothalamic–pituitary–adrenal (HPA) axis dysfunction theory, based on increased resistance to glucocorticoids and resulting cortisol hypersecretion; the neurogenesis hypothesis, based on a lack of adult neuronal growth leading to mood changes; and the neuroinflammatory or macrophage/cytokine theory, based on external psychosocial stressors and internal organic inflammatory conditions causing depression via increased inflammatory cytokine production (Smith, 1991; Maes, 1993, 1995; Maes et al., 2009).
Inflammatory cytokines that enter the brain can affect a number of neural pathways including those involved with mood. The cytokines may alter the abovementioned pathways to cause depressive symptoms. A number of correlative studies suggest a role for inflammation in the pathophysiology depression. The most consistent observations include:
Higher rates of MDD diagnosis in persons with inflammatory medical illnesses (Dickens et al., 2002; Graff et al., 2009; Lo Fermo et al., 2010; Pascoe et al., 2011; Krishnadas and Cavanagh, 2012).
Higher levels of inflammatory markers, acute phase proteins, and cytokines such as C-reactive protein, tumour necrosis factor α (TNF-α), interleukin-6 (IL-6), interleukin-1 (IL-1), acute phase reactant proteins, and prostaglandins in the serum and cerebral spinal fluid of individuals with MDD in the absence of any other medical condition (Maes, 1993; Song et al., 1994; Seidel et al., 1995; Sluzewska et al., 1996; Dowlati et al., 2010; Liu et al., 2011).
Patients undergoing treatment with cytokines (interferon α, IFN-α) and IL-2 experience more frequent and more severe depressive symptoms than individuals with the medical conditions prior to commencing cytokine therapy (Bonaccorso et al., 2001; Raison et al., 2008; Myint et al., 2009; Krishnadas and Cavanagh, 2012;).
Multiple classes of antidepressants have been associated with a negative immunoregulatory effect through inhibition of pro-inflammatory cytokines and stimulation of anti-inflammatory cytokines. It has been suggested that the therapeutic efficacy of antidepressants may be due to a negative immunoregulatory mechanism (Xia et al., 1996; Maes et al., 1999; Kubera et al., 2001).
Baune et al. (2012) in the Sydney Memory and Aging Study suggested that cytokines may be aetiological factors in depression. They may be used as markers for depressive symptoms at differing stages of the disease in elderly patients. Their study is the first to report that the chemokine IL-8 is associated with depressive symptom severity at baseline and 2-year follow up (Baune et al., 2012).
In patients with known inflammatory medical conditions, the prevalence of MDD is 5–10-times higher than that of the general population and these individuals tend to have a worse prognosis with a higher disability rate (Krishnadas and Cavanagh, 2012). MDD is also associated with central nervous system (CNS) inflammatory conditions, for example people with multiple sclerosis have exhibited a prevalence rate of up to 50% after 14-year follow up (Lo Fermo et al., 2010). Additionally, a recent systematic review of post-stroke depression reported a conservative prevalence rate of 30% in surviving patients as discussed later (Pascoe et al., 2011). The review implicated inflammation-induced cell death in mood-related brain regions as an explanation for post-stroke depression (Pascoe et al., 2011). MDD is also associated with peripheral inflammatory conditions such as psoriasis, rheumatoid arthritis, and inflammatory bowel disease, with conservative estimated prevalence rates between 13 and 17% (Dickens et al., 2002; Graff et al., 2009; Lo Fermo et al., 2010; Krishnadas et al., 2011; Pascoe et al., 2011).
Inflammatory conditions may not be a cause of depression but may rather be part of a generalized physiological response to an underlying dysregulation in the immune system. For example, the presence of MDD was the only psychosocial variable which independently predicted worse short-term remission and increased negative long-term outcomes in patients with Crohn’s disease treated with infliximab (Persoons et al., 2005). The authors study suggested this may be due to an underlying immune dysregulation and enhanced cytokine production in patients with MDD (Persoons et al., 2005).
This review summarizes recent research on the relationship between inflammatory cytokines and depression via: (1) alterations in neuroendocrine function; (2) changes in neurotransmission pathways; and (3) via neuroprogression which is defined as the combined effect of neurotrophin depletion, neurodegeneration, neuronal apoptosis, decreased neurogenesis, and neuroplasticity on the development of depression (Berk et al., 2011). Figure 1 summarizes the pathways discussed in the article and how they may inter-relate in the pathogenesis of depression.
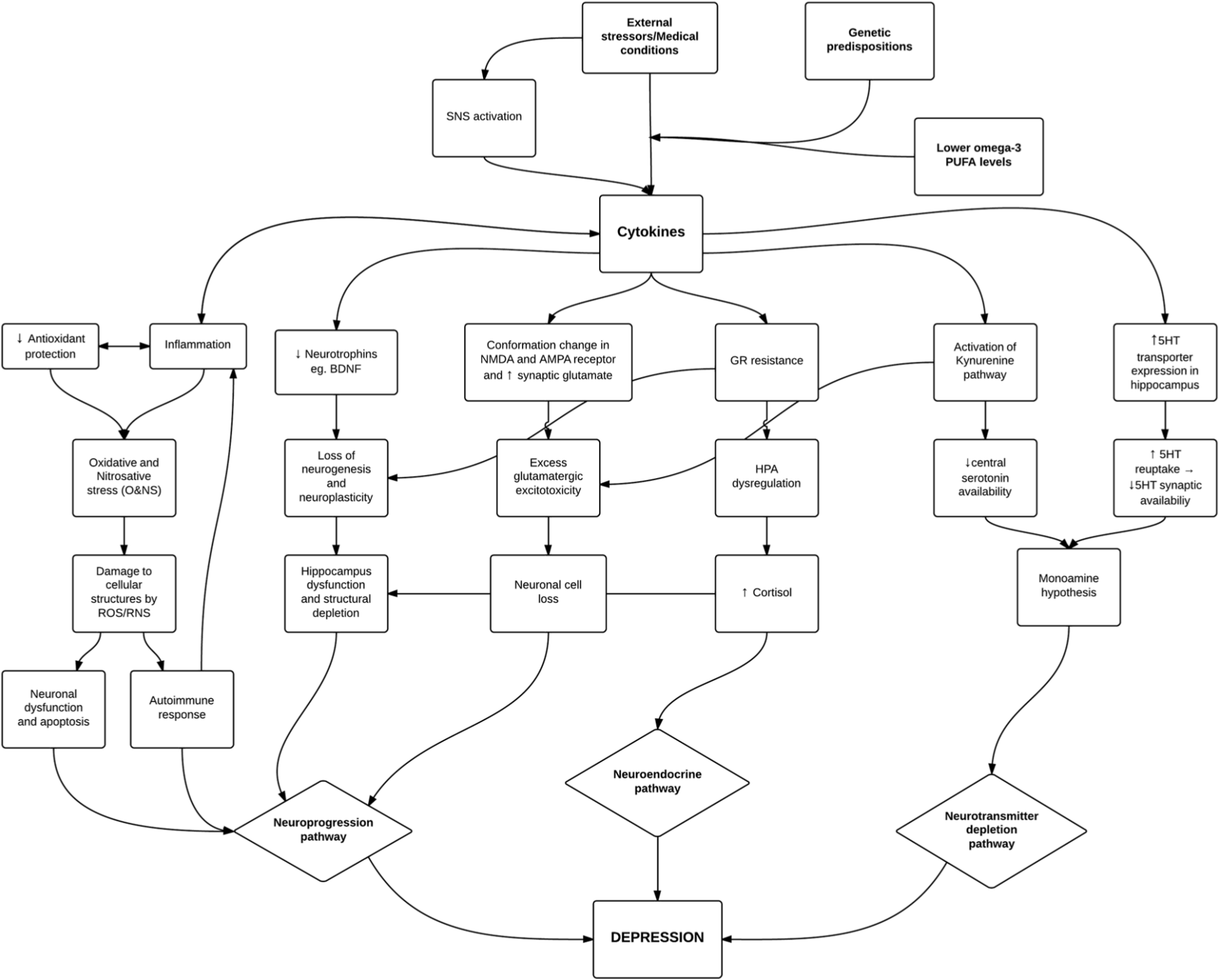
Inflammatory pathways involved in the pathogenesis of depression. Genetic predispositions and lower PUFA levels may potentiate the effect of external stressors and medical illnesses to influence increased cytokine production either centrally or peripherally. Cytokines have a central role by acting via various pathways outlined above and affect neuroprogression, neuroendocrine and neurotransmitter depletion pathways. There are multiple interactions between these pathways suggesting the existence of a complex model for the development of depression. PUFA = polyunsaturated fatty acid, SNS = sympathetic nervous system, CNS = central nervous system, GR = glucocorticoid receptor, BDNF = brain derived neurotrophic factor, 5HT = serotonin, HPA = hypothalamic-pituitary-adrenal.
Methods
An online literature search was carried out online in July 2012 using PubMed. Combinations of different search terms were employed to ensure maximum results; the terms used included “major depressive disorder”, “depression”, “inflammation”, “inflammatory”, “immune”, “cytokines”, “hypothalamic-pituitary-axis”, “oxidative and nitrosative stress”, and “antioxidant” and “omega-3”. First, the titles of all the results were read to select articles which relevant to the topic of this review. Second, the abstracts of the selected articles were reviewed to further identify articles that were appropriate, and the full texts of these articles were collected for analysis. The reference sections of all the full text articles were reviewed to collect additional articles that were missed in initial search. Original research and reviews were included to maximize the selection of information included in this review. Additional content was included in January 2013 drawing on new literature relevant to this review.
Results
Cytokines in the brain
Pro-inflammatory cytokines may affect CNS neural circuits to cause a cascade of events that contribute to the development of major depression. Until recently the CNS was thought to be restricted from the immune system. However it has been observed that cytokines may enter the CNS by de-novo synthesis by activated neurons, microglia, and astrocytes, or indirectly via the blood brain barrier (BBB) (Krishnadas and Cavanagh, 2012). These immunotransmitters are produced centrally in response to CNS and peripheral inflammation, as seen in autoimmune multiple sclerosis, or secondary to an ischaemic insult as seen in post-stroke patients (Raison et al., 2008; Krishnadas and Cavanagh, 2012).
Individuals on exogenous cytokine therapy have increased endogenous cytokine production and this is associated with the development of depressive symptoms, with prevalence rates between 20 and 30% (Bonaccorso et al., 2001; Myint et al., 2009; Karg et al., 2011). Patients with hepatitis C treated with IFN-α have increased cerebral spinal fluid levels of IFN-α and IL-6 and these were correlated with the concentration of administered IFN-α, but not peripheral levels of these cytokines. This suggests that exogenous IFN-α caused de-novo synthesis of these immunotransmitters (Raison et al., 2008). The study also found reduced serotonin metabolite, 5-hydroxyindoleacetic acid (5-HIAA), which was the primary predictor of depressive symptoms. IL-6 levels may have altered serotonin metabolism, thereby accounting for the depressive phenotype (Raison et al., 2008). It must be noted that the manifestation of these depressive symptoms, whilst similar to MDD, is a part of a “sickness behaviour” seen in patients on cytokine therapy and not that of psychiatric MDD.
Peripheral cytokines can also signal the brain by gaining access through the BBB and consequently affect neural pathways. They penetrate the BBB by different mechanisms, some of which include active transport, passive passage through “leaky” regions in the BBB, and transmission along afferent vagal pathways (Miller et al., 2009a; Capuron and Miller, 2011; Krishnadas and Cavanagh, 2012). In two separate trials, healthy volunteers were injected with lipopolysaccharide and Salmonella typhi vaccine; these individuals developed symptoms of depression and anxiety and the severity correlated with increases in peripheral cytokine levels (Reichenbert et al., 2001; Brydon et al., 2008). Once cytokines enter the brain, through primary production or secondary entry, they have the ability to affect central pathways that may lead to development of major depression (Miller et al., 2009a).
Higher rates of comorbid depression in infectious, autoimmune, and neurodegenerative diseases are not sufficiently explained by the psychological distress of the starting disease (Pollak and Yirmiya, 2002). Therefore, the role of biological mechanisms like pro-inflammatory cytokines cannot be discounted completely in the aetiopathogenesis of depression.
Omega-3 polyunsaturated fatty acids (PUFA) depletion may be associated with depression due to a loss of their anti-inflammatory effect. Epidemiological studies showed that lower dietary intakes of omega-3 PUFA are associated with higher rates of depression (Hibbeln, 1998; Leonard and Maes, 2012). Supplementation with eicosapentaenoic acid, an omega-3 PUFA, in depression showed significant antidepressant activity (Lin and Su, 2007). A possible mechanism for the antidepressant effect of omega-3 lies in its ability to attenuate proinflammatory cytokine production. Eicosapentaenoic acid reduces the synthesis of prostaglandin E2, IL-1, IL-6, TNF-α, and IFN-γ (Leonard and Maes, 2012). Subjects with low omega-3 PUFA levels show significantly higher stress-induced production of proinflammatory cytokines compared to subjects with higher levels of omega-3 PUFA, and these are associated with higher anxiety and perceived stress ratings (Leonard and Maes, 2012).
Neuroendocrine disturbances, inflammation, and depression
HPA axis dysregulation in depression
Since the 1970s, it has been noted that up to 50% of patients with depression have raised serum cortisol and dexamethasone non-suppression (Carroll, 1982). Depressive symptoms are a frequent side effect of glucocorticoid treatment and a symptom of Cushing’s syndrome (Zunszain et al., 2011). Normally, the HPA axis is appropriately activated in the body’s response to environmental stress, but some depressed patients appear to have an abnormal negative feedback system in the setting of intact pituitary and adrenal sensitivity leading to excessive cortisol secretion (Lopez-Duran et al., 2009). Chronic hyperactivity may result in long-lasting problems and may explain some of the symptoms of MDD. There are a growing number of correlative studies that demonstrate elevated levels of both glucocorticoids and inflammatory cytokines in depressed individuals. When this is combined with communication mechanisms that exist between the endocrine, immune, and central nervous systems, it gives rise to the hypothesis that inflammatory mechanisms may affect neuroendocrine processes to cause depression (Zunszain et al., 2011).
Some patients with MDD have raised corticotrophin-releasing hormone and its mRNA, which have been associated with multiple depressive symptoms including fear/anxiety, changes in sleep patterns, altering locomotor activity, and food intake (Pace and Miller, 2009). Furthermore, hypercortisolism has been associated with increased presence of distress symptoms and maladaptive coping styles, suggesting it may have a role in depression spectrum disorders (Kunugi et al., 2012).
Glucocorticoid mechanism of action
The mechanism of glucocorticoid action is to bind to cytosolic glucocorticoid receptor (GR), cause a conformational change in GR, resulting in its dissociation from inactivating chaperone proteins and subsequent translocation into the nucleus. Once in the nucleus, GR acts directly as a transcription factor to express anti-inflammatory genes, or act indirectly with other co-repressor molecules to inactivate inflammatory signalling pathways, for example nuclear factor-kappa B (NF-κB). Reduction in GR function may dysregulate the HPA axis and is hypothesized to contribute to depressive symptoms by a loss of inhibition of the inflammatory pathways (Zunszain et al., 2011).
Cytokines and HPA axis dysregulation
Cytokines can cause hyperactivity of the HPA axis, with subsequent elevated systemic cortisol levels (Maes et al., 1993b; Zunszain et al., 2011). Cytokines cause GR resistance to cortisol and result in loss of the negative feedback over the HPA axis (Pace and Miller, 2009; Krishnadas and Cavanagh, 2012). Several molecular mechanisms have been identified to explain how cytokines can cause a functional GR resistance. IL-6 and TNF-α prevent the entry of the cortisol-GR complex into the nucleus of neurons. They also prevent the binding of the cortisol-GR complex to the DNA and inhibit DNA transcription (Pace et al., 2011). The cytokines act on pathways such as the MAPK, NF-κB, signal transducers and activators of transcription, and cyclooxygenase, and inhibit GR translocation into the nucleus and GR-mediated gene transcription, and reduce expression of intracellular GR (Zunszain et al., 2011; Krishnadas and Cavanagh, 2012). The result of inflammatory cytokines’ action on the HPA axis is functional glucocorticoid resistance resulting in steroid insensitivity and cortisol hypersecretion (Zunszain et al., 2011; Schmidt et al., 2011). Restoration of the HPA axis abnormalities has been associated with clinical response to treatment (Zunszain et al., 2011; Kunugi et al., 2012).
There may be a cyclic crosstalk between cytokines and glucocorticoids, implying the HPA axis hyperactivity and inflammation may be part of a common pathophysiological response.HPA axis hyperactivity is a marker of GR resistance and steroid insensitivity on target tissues, which may lead to a loss of the glucocorticoid anti-inflammatory effect; also, inflammatory cytokines can cause HPA axis hyperactivity directly or by inducing GR resistance (Zunszain et al., 2011). Zunszain et al. (2011) proposeed that elevated cytokines in the presence of glucocorticoid resistance may be a contributor to depressive symptoms, as opposed either process occurring in isolation. They suggested that chronic stressors cause persistent hypercortisolaemia which causes immune cells to undergo a compensatory downregulation of GR activity. This limits the ability of cortisol to inhibit the immune response even in the presence of high circulating cortisol levels. The result is a chronic low-grade inflammatory state which has been associated with infectious and autoimmune diseases as well as depression (Zunszain et al., 2011). There is also evidence in animal and human studies that administration of pro-inflammatory cytokines leads to behavioural changes that correlate to a depressive phenotype, including depressed mood, fatigue, disrupted sleep, anxiety, and suicidal ideation (Raison et al., 2006; Pace and Miller, 2009).
Neurotransmitters, inflammation, and depression
The monoamine hypothesis
The monoamine hypothesis states that a central synaptic deficiency of serotonin and/or noradrenaline cause depressive symptoms. It is believed that there is a reduction in serotonin and noradrenaline neurotransmission from their midbrain nuclei in the raphae nuclei and locus coeruleus, into the limbic, prefrontal cortex, and hippocampus (Palazidou, 2012).
Cytokines and reduced serotonin synthesis
Inflammatory cytokines can alter the synthesis and reuptake of central mood modifying neurotransmitters such as serotonin (Miller and Timmie, 2008). Cytokines may lower central serotonin levels by altering tryptophan (a precursor for serotonin synthesis) metabolism. Maes et al. (1993a) found lower plasma trypthophan levels in patients with major depression and hypothesized that this may be due to an immune response. IL-1β and TNF-α induce indoleamine 2,3-dioxygenase (IDO), an enzyme which activates the kynurenine pathway. The cytokines act via various inflammatory signalling cascades, such as the signal transducer and activator of transcription 1a, interferon regulatory factor-1, NF-κB, and p38 mitogen activated protein kinase (MAPK) pathways (Miller et al., 2009a; Zunszain et al., 2011; Krishnadas and Cavanagh, 2012). The kynurenine pathway is responsible for metabolizing dietary tryptophan away from serotonin (5HT) synthesis, and redirecting it to produce other metabolites, namely, kynurenine, 3-hydroxy-kynurenine, and quininolinic acid. The result is a reduction in serotonin synthesis, and subsequent central serotonin deficiency, and the potentiation of depressive symptoms (Pace and Miller, 2009; Krishnadas and Cavanagh, 2012). Some studies suggest kynurenine and quininolinic acid affect mood, independent of their effect on serotonin, as discussed later (Miller et al., 2009a).
Cytokines and increased serotonin reuptake
Selective serotonin reuptake inhibitors act by inhibiting serotonin reuptake from neural synapses and this is a mechanism by which they are thought to have their antidepressant effect. Cytokines oppose this effect by stimulating increased serotonin reuptake from synapses. Increased cerebral spinal fluid IL-6 levels are capable of activating the IDO pathway, as discussed above, and also the MAPK pathway. The MAPK pathway increases the activity of cell membrane transporters for serotonin, dopamine, and norepinephrine in the rat brain, causing increased reuptake of these neurotransmitters (Miller et al., 2009a). IL-1 and TNF-α have also been associated with activation of the MAPK pathway and upregulation of serotonin transporters in the hippocampus (Bufalino et al., 2012). Cytokine activation of the IDO and MAPK pathways deplete synaptic serotonin levels by reducing their synthesis and increasing reuptake respectively.
Depletion of neurotrophic factors in depression
Brain volumetric studies have shown decreased volumes in regions of the brain associated with mood. It is believed that these changes are due to lower levels of neurotrophic factors that reduce neuroplasticity and neurogenesis and therefore protection against depression. External stressors downregulate the production of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and VGF nerve growth factor (Kubera et al., 2011). BDNF is a neurotransmitter that plays a major role in neuronal growth, survival, maturation, and synaptic plasticity in the adult hippocampus (Palazidou, 2012). VGF has a role in synaptic plasticity to reverse depressive-like behaviour and enhance hippocampus proliferation (Thakker-Varia et al., 2007). Low serum BDNF levels have been reported in depressed individuals and appear to correlate with severity of disease (Palazidou, 2012). Chronic stress reduces BDNF synthesis by causing demethylation of histones at the BDNF promoter region (Tsankova et al., 2006). This is supported by findings of decreased BDNF concentrations in response to stress, particularly in the limbic regions that mediate mood (Shimizu et al., 2003). Another neurotrophic factor is fibroblast growth factor ; reduced activity in the this system can also alter brain development and predispose an individual to develop depression (Turner et al., 2006).
Antidepressant therapy exhibits neurogenic effects by stimulating the production of neurotrophic factors such as BDNF, the receptor for BDNF trkB, and neural growth factor (Kubera et al., 2011). This results in increased neurogenic activity in the hippocampus and the prefrontal cortex, giving rise to improving cognitive flexibility and subsequent increase in ability to cope with environmental challenges that may otherwise potentiate a depressive episode (Schmidt et al., 2011). These studies suggest that reduced neurotrophic factor levels may contribute to depressive symptoms and that antidepressant treatment reverses these symptoms by restoring neurotrophic factor activity. These studies support one part of the neuroprogression hypothesis model of depression, as defined earlier.
Preliminary data shows that administration of IL-1β receptor antagonist into the hippocampus of mice blocks stress-induced BDNF depletion (Barrientos et al., 2003). IL-1 and TNF-α decrease hippocampal expression of BDNF and its receptor, resulting in decreased hippocampal neurogenesis (Wu et al., 2007; Miller et al., 2009a). These finding suggests that inflammatory mechanisms can reduce BDNF levels and potentiate depressive symptoms via altered neurogenesis.
Neuroprogression, depression, and inflammation
There is evidence that neuroprogression mechanisms underlie the development of depression. The previous section addressed how neuroptrophin factor depletion is associated with depression; the following sections outline some of the other key mechanisms involved in neuroprogression, including neurodegeneration, neuronal apoptosis, decreased neurogenesis, and neuroplasticity.
Neurogenic hypothesis of depression
According to the neurogenic hypothesis of depression, new neuronal connections are needed in the adult brain for adequate mood control and antidepressant efficacy (Petrik et al., 2012). Research into this hypothesis has gained interest because of a number of correlative studies that showed that humans with depression have decreased volumes of selective brain regions, because of decreased adult neurogenesis and increased neurodegeneration (Maes et al., 2009; Eisch and Petrik, 2012). Some of the areas in the brain affected include the anterior cingulate cortex, orbitofrontal cortex, and the hippocampus (Koolschijn et al., 2009). The hippocampus is involved in learning/memory context-dependent emotional responses (Fanselow, 2000), and mood control (Petrik et al., 2012). It is because of its role in mood control that the hippocampus has been the subject of depression research. Interestingly, hippocampal volume is reduced in patients with multiple episodes of depression but not with first-episode depressive symptoms (MacQueen et al., 2003; Palazidou, 2012). This implies that hippocampal dysfunction occur prior to any detectable structural changes on imaging studies and that repeat episodes cause additive damage which eventually becomes grossly visible with chronicity.
Neurogenic-neuroendocrine interaction
Eisch and Petrik (2012) recently coined the term “neurogenic interactome” to describe a complex series of endocrine and neurochemical cascades, and reciprocal connections between brain regions which influence adult neurogenesis and have downstream effects on behaviour (Eisch and Petrik, 2012). They describe how intact neurogenesis in the hippocampus is key to inhibition of the hypothalamus and regulation of the HPA axis. This contributes to mood control and preventing the development of depressive symptoms (Eisch and Petrik, 2012; Petrik et al., 2012).
The functional inhibition of the hippocampus on the HPA axis is supported by the richness of corticosteroid receptors in the hippocampus (Reul and deKloet, 1986) and its anatomical link to the hypothalamus via the fornix (Palazidou, 2012). This inhibitory control is lost after damage to the hippocampus. There is resultant HPA axis dysfunction, leading to the cognitive and emotional symptoms of depression (Zunszain et al., 2011). This disinhibition of the HPA access may be due to impaired adult neurogenesis in the hippocampus. Animal models reveal how chronic stress (a surrogate for psychosocial precipitants for depression) causes raised glucocorticoid levels and hippocampal shrinkage manifested by dendritic retraction, suppression of adult neurogenesis, and increased neuronal cell death (Czeh and Lucassen, 2007).
Persistent hypercortisolaemia acts via voltage-gated ion channels facilitating calcium entry into neurons to further cause neuronal cell death (Palazidou, 2012). The antidepressant sertraline modulates GR expression and function, and this has been associated with enhanced neurogenesis (Anacker et al., 2011). Trials with GR antagonist mifepristone have shown relief of symptoms in psychotic depression after 4–8 days of treatment (Belanoff et al., 2002). In the rat brain, mifepristone reversed corticosterone-induced loss in neurogenesis and survival (Mayer et al., 2006). This may be the mechanism by which mifepristone relieved depressive symptoms in humans. These findings support the hypothesis that persistent hypercortisolaemia cause depressive symptoms by interacting with GR in the hippocampus to cause a loss of neurogenesis and increased cell death.
Inflammation and the neurogenic theory of depression
If the neurogenic hypothesis of depression is a valid hypothesis for the development of major depression, then the next question is, “What inflammatory mechanisms inhibit neurogenesis and thereby lead to depressive phenotypes?” Inflammatory changes have been implicated in neuronal cell death via excitotoxic mechanisms. Excess glutamate signalling induces neuron excitotoxicity, resulting in structural changes in the brain that have been studied in disorders such as stroke, neurodegenerative diseases, and, most recently, depression (Lee et al., 2002). IL-1β binds to the IL-1 R1 receptor in the hippocampal neurons and induces phosphorylation of the N-methyl-
The kynurenine pathway byproducts 3-hydroxy-kynurenine and quininolinic acid, contribute to neurotoxicity and neurodegeneration (Christmas et al., 2011). 3-Hydroxy-kynurenine causes increased oxidative stress and contributes to neuron apoptosis. Quininolinic acid causes oxidative stress and is an NMDA receptor agonist contributing to excitotoxic neurotoxicity (Myint and Kim, 2003). Additionally, TNF-α leads to a change in the conformation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor, facilitating calcium influx into the neuron, and consequent glutamate activated excitotoxicity (Stellwagen et al., 2005).
Inflammatory cytokines inhibit neurogenesis by causing cell death directly via excitotoxic mechanisms and apoptosis in the hippocampus, and indirectly via interaction with neuroendocrine pathways, namely hypercortisolaemia.
The oxidative and nitrosative stress (O&NS) pathways
A new hypothesis centres around the generation of free radicals in the setting of antioxidant scarcity, leading to further neuroprogression and depression. Inflammation and mitochondrial metabolism generate highly reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS/RNS may cause damage to fatty acids, membrane lipids (lipid peroxidation), DNA, proteins, and mitochondria, with consequent cellular dysfunction, apoptosis, and tissue destruction (Maes et al., 2011). There is evidence that proinflammatory cytokines such as IL-1 and TNF-α potentiate the effects of free radicals. The implication is that cytokines may stimulate the neuroprogression of depression via the O&NS pathway (Maes et al., 2012).
Normally, free radicals are balanced by protective antioxidants, antioxidant enzymes, and proteins. Examples of antioxidants include coenzyme Q10, vitamins C and E, and glutathione. Antioxidant enzymes include superoxide dismutase (SOD) and glutathione peroxidase and proteins include albumin, transferrin, haptoglobins, and ceruloplasmin. A lowered antioxidant capacity impairs protection against the free radicals and allows for the damage to cellular structures. Organs like the brain are particularly vulnerable to the O&NS neuronal damage and neurodegeneration because of a high metabolic rate and lower antioxidant levels (Maes et al., 2011).
Oxidative stress can alter the immunogenicity of ubiquitous cellular products to induce an autoimmune response. O&NS pathways may change the chemical structures of innate molecules to create neoepitopes that are highly immunogenic. The result is an immunoglobulin mediated autoimmune response against the fatty acids and protein neoepitopes (Maes et al., 2011; Leonard and Maes, 2012).
Current evidence supports the O&NS hypothesis as depression is associated with decreased antioxidant levels, increased O&NS activity, and increased cellular damage and autoimmune response secondary to O&NS. In depression, there is a significant decrease in antioxidant levels such as tryptophan, tyrosine, albumin, zinc, vitamin E, and glutathione (Maes et al., 2000). Many studies show an increased level of ROS and RNS production in depression; however, the values are higher in the acute phase of depression and normalize when the depression becomes more chronic (more than 2 years duration) (Maes et al., 2011)
The decrease in antioxidant levels and increase free radical result in oxidative damage and apoptosis and may explain the volumetric changes in the brain of depressed subjects. Malondialdehyde (MDA) is a byproduct of polyunsaturated fatty acid peroxidation and is used as a measure for lipid peroxidation and oxidative stress. Multiple studies have found increased MDA levels in the blood of people with depressions compared with healthy subjects (Ozcan et al., 2004; Galecki et al., 2009). These levels were reduced with treatment with antidepressants. There is increased oxidative damage to DNA by ROS in depression, as the molecular marker for DNA damage is significantly increased in patients with recurrent depressive episodes (Forlenza and Miller, 2006).
There is evidence that depression is associated with increased autoimmune response to neoepitopes generated secondary to O&NS pathways. Depressed subjects have raised plasma IgG autoantibodies against low-density lipoproteins and IgM-mediated immune responses against phosphatidylinositol (PI) as compared to healthy subjects. The IgM-mediated response against PI was significantly correlated to symptoms of depression such as sadness and fatigue (Maes et al., 2007; Leonard and Maes, 2012).
There is evidence that antidepressants can counter the effects of O&NS by having an antioxidant-like effect and this may be another way they achieve their therapeutic effect (Maes et al., 2011; Leonard and Maes, 2012). There are numerous hypotheses with growing evidence to support the idea that depression is a result of neuroprogression. The various pathways that lead to neuroprogression are closely linked to inflammation and proinflammatory cytokines. The most recent O&NS pathway highlights how inflammation generates free radicals that may cause neuroprogression via two main mechanisms; directly by cellular destruction and indirectly by the inhibition of immune tolerance and activation of autoimmune mechanisms.
Genetics
Some studies have implicated the serotonin transporter gene (SLC6A4) as a common gene affecting both depression and immune function. However, there are conflicting findings linking polymorphisms in this gene to predispose individuals to depressive symptoms. Su et al. (2009) reported that participants with a specific haplotype at SLC6A4 had both increased depressive symptoms and elevated plasma IL-6; however, they acknowledged that other studies reported no significant differences between depressive symptoms and polymorphisms of SLC6A4 (Su et al., 2009).
Polymorphisms in the human serotonin transporter gene promoter region (5-HTTLPR), leading to the “short” (SS) allele, have been associated with an increased likelihood or vulnerability to develop depression after stressful life events (Caspi et al., 2003), chronic illness (Otte et al., 2007), or IFN-α administration (Bull et al., 2009).
One study analysed cytokine concentrations in healthy individuals with and without the SS allele to determine any variance which may account for the predisposition to depression (Fredricks et al., 2010). They found that healthy individuals with the SS polymorphism had a higher IL-6/IL-10 ratio at baseline and after stress testing. This was suggestive of a chronic proinflammatory state under both resting and stressful situations. The authors hypothesize that these genetic variations cause a chronic proinflammatory state and thereby increase these individuals’ vulnerability to develop depression, acting as a predisposing factor (Fredricks et al., 2010).
Studies investigating the relationship of IL-1β gene polymorphisms and depression have reported inconsistent results. Early studies associate depression with the T/T genotype that causes higher IL-1β concentrations (Rosa et al., 2004); however, later studies linked the low IL-1β producer, C/C genotype, with depressive symptom severity (Hwang et al., 2009). Yet another study found that a combination of the C and T alleles at different locations in the gene was associated with recurrent major depressive episodes via enhanced binding to transcription factors, resulting in increased production of IL-1β (Bufalino et al., 2012). Additionally, patients with specific polymorphisms of the IL-1β gene were less likely to respond to antidepressant treatment, supporting the idea that immune genes are important not only in the aetiology of depression, but also in efficacy of treatment (Su et al., 2010).
Polymorphisms in the enzymes phospholipase-A2 (PLA2) and cyclooxygenase-2 (COX2) were associated with increased risk of developing IFN-α-induced depression in patients with hepatitis C. They found that polymorphisms in these enzymes led to a reduction in polyunsaturated fatty acids (PUFAs) docohasexaenoic acid and eicosapentaenoic acid, which are believed to play a protective role in depression (Su et al., 2010). These genetic polymorphism and reduction in PUFAs rendered individuals 3-times more likely to develop IFN-α-induced depression than those without these genetic polymorphisms (Su et al., 2010).
The degree of inconsistency and lack of reproducibility of the genetic links between inflammation and depression warrants further research in this field. Depression, like hypertension and diabetes, is a complex disorder involving environmental factors that interact with genetic predispositions and is likely to involve multiple genes and unlikely to be the result of a defect in any single gene (Bloch and Singh, 2007). The discovery of a set of genes which may predispose an individual to developing depression or resistance to treatment would be clinically valuable as these individuals may benefit from tailored prophylactic treatment (Bufalino et al., 2012).
Psychosocial stressors and inflammatory cytokines
Psychosocial stress can directly activate peripheral and central inflammatory cascades which may act as pathways to trigger or perpetuate depressive symptoms. Individual healthy volunteers exposed to public speaking and other stressors were found to exhibit increased DNA binding of the chief inflammatory transcription factor NF-κB in peripheral mononuclear cells (Bierhaus et al., 2003). NF-κB and IL-6 response to psychosocial stressors are exaggerated in patients with depression as compared to non-depressed individuals (Pace et al., 2006). Psychosocial stressors may also induce O&NS pathways directly as evidenced in men and animal studies (Leonard and Maes, 2012). Stress-induced production of these CNS cytokines is mediated through microglia (Frank et al., 2007). It may also be due to activation of the sympathetic nervous system, which is active during acute stress. Stress-induced catecholamines have been shown to increase cytokine expression in the brain of rats (Johnson et al., 2005). Alpha-adrenergic antagonists can block stress-induced rises in peripheral IL-6 levels in humans, suggesting a possible link between psychosocial stress leading to SNS activation, cytokine production, and the resulting depressive symptoms (Mazzeo et al., 2001).
Potential applications
A number of retrospective studies report depressed patients with poor response to conventional antidepressant treatment were more likely to have raised inflammatory markers, TNF-α and IL-6, prior to treatment (Maes et al., 1997; Miller et al., 2009b). Schmidt et al. (2011) suggested the use of a panel of biomarkers, including inflammatory molecules to diagnose, create biological subtypes of depression and predict response to treatment in order to optimize clinical outcomes. There is also some evidence for the benefit of adjuvant pharmacological drugs, such as the addition of celecoxib (a COX2 inhibitor) to reboxetine and fluoxetine to achieving higher rates of improvement in depressive symptoms than the respective antidepressants alone (Muller et al., 2006). Similar findings were reported in psoriasis patients receiving the TNF-α antagonist entercept; they experienced greater improvements in depressive symptoms compared with placebo-treated patients and this difference was independent of any improvement in psoriasis activity (Tyring et al., 2006). Evidence analysed in this review supports further research may be warranted to biologically subtype depression based on variations in inflammatory markers.
Discussion
There is a large and growing body of evidence for the role of inflammatory mechanisms interacting with neural pathways involved in mood, which may underlie the development of depressive symptoms. However, it is important to appreciate that the majority of people with inflammatory conditions do not suffer from depression, and the majority of people with depression do not have inflammatory conditions. This serves to remind us that inflammation alone does not cause depression and is certainly not necessary for depression to occur. Also, if depression were an inflammatory illness, it may be reasonable to hypothesize that anti-inflammatory drugs such as non-steroidal anti-inflammatory drugs (NSAIDs) or steroids may alleviate depressive symptoms; however, there is evidence that NSAIDs actually attenuate the antidepressant response to selective serotonin reuptake inhibitors (Gallagher et al., 2012).
The question that arises is, “What role does inflammation actually play in depression?” Krishnadas and Cavanagh (2012) suggest that inflammation may act as a trigger in a cascade of events that culminates in a depressive phenotype. Raison and Miller (2011) propose that inflammation contributes to depression in only a subset of patients and that inflammation is not an absolute “depressogenic” process. They suggest the notion of a “super-network” with immune response amplification, constituting of different mechanisms through which inflammation may act to precipitate a depressive phenotype. Some of these mechanisms include glucocorticoid insensitivity, reduced parasympathetic signalling, and reduced BDNF levels (Raison and Miller, 2011; Krishnadas and Cavanagh, 2012). Inflammatory processes may affect a combination of pathways in a subset of susceptible patients to trigger the development of a depressive phenotype.
Many studies at present are correlative in nature; however, correlation is not causation. Further research is needed to demonstrate a causative link between inflammatory processes and depression.
Conclusion
The role of inflammation in the aetiopathogenesis of depression may be conceptualized in terms of a psychiatric formulation focused on the biological model of disease, divided into predisposing, precipitating, perpetuating, and protective factors that all add up to cause depression. Various genetic polymorphisms and a chronic pro-inflammatory state may predispose susceptible individuals to develop depression as well as predict a failure to respond to treatment. Cytokines affect three major neural pathways involved in the aetiopathogenesis of depression. These disturbances may precipitate and perpetuate and a loss in neuroprotective mechanisms that all eventuate in the development of major depression. These mechanisms include: the neuroendocrine pathway, by causing HPA axis dysregulation and cortisol hypersecretion; the neurotransmitter pathway, by reducing synaptic serotonin availability and depleting central neurotrophin levels to reduce its neuroprotective effect and loss of neurogenesis; and the neuroprogression pathway, by causing a loss of hippocampal neurogenesis and volume which in turn causes more HPA axis dysregulation. External and/or internal stressors induce proinflammatory cytokine production which in turn precipitates and perpetuates depressive symptoms via their action on multiple neuronal and neuroendocrine pathways. Despite the large body of correlative and experimental research implicating inflammation in the aetiopathogenesis of depression, more research is needed to determine the extent to which inflammatory cytokines contribute to depressive symptoms and exact the mechanisms by which these occur. Currently, there is still no consensus on a pathophysiological model of depression, but rather the existence of multiple observations and empirical data that provide, at best, hypotheses on the mechanisms underlying this complex disease. By understanding these mechanisms further, in the future, we may be able to create biological subtypes of depression and tailor treatment more optimally for patients with depression.
Footnotes
Acknowledgements
We would like to thank Dr Jason Lee for his advice regarding this review.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.