
Abstract
Objective:
There is considerable evidence to suggest that the abuse of illicit drugs, particularly cannabis and methamphetamine, has aetiological roles in the pathogenesis of psychosis and schizophrenia. Factors that may increase susceptibility to the propsychotic effects of these drugs include the age at which the abuse starts as well as family history of genetic polymorphisms relevant to the pathophysiology of this disorder. However, the neurobiological mechanisms involved in drug abuse-associated psychosis remain largely unclear.
Methods and Results:
This paper presents an overview of the available evidence, including clinical, animal model, and molecular studies, with a focus on brain regions and neurotransmitters systems, such as dopamine and glutamate, previously implicated in psychosis.
Conclusion:
It is clear that further studies are urgently needed to provide a greater insight into the mechanisms that mediate the long-term and neurodevelopmental effects of cannabis and methamphetamine. A dialogue between basic science and clinical research may help to identify at-risk individuals and novel pathways for treatment and prevention.
Introduction
Schizophrenia is a highly heterogeneous disorder with no single cause. Numerous genetic, developmental, and environmental risk factors are known to be involved and it is possible that they may cause different pathophysiological processes that ultimately produce similar clinical endpoints. One of the predominant environmental factors suggested by epidemiological, clinical, and basic science studies is the abuse of drugs such as methamphetamine and cannabis (American Psychiatric Association, 2000; National Centre for Classification in Health, 2008). According to a recent review, cannabis is the drug of choice with between 149 and 271 million users worldwide and this is followed closely by amphetamines (Degenhardt and Hall, 2012). One of the key differences between the two drugs of abuse is that cannabis abuse appears to be able to cause psychosis as well as other symptoms that fit the diagnostic classification for schizophrenia, whereas methamphetamine abuse appears to be associated with psychosis only (American Psychiatric Association, 2000). The purpose of this review is to discuss the findings of clinical and preclinical studies that have investigated the significance of cannabis or methamphetamine abuse as a risk factor for the development of psychosis.
Human studies
Cannabis and schizophrenia
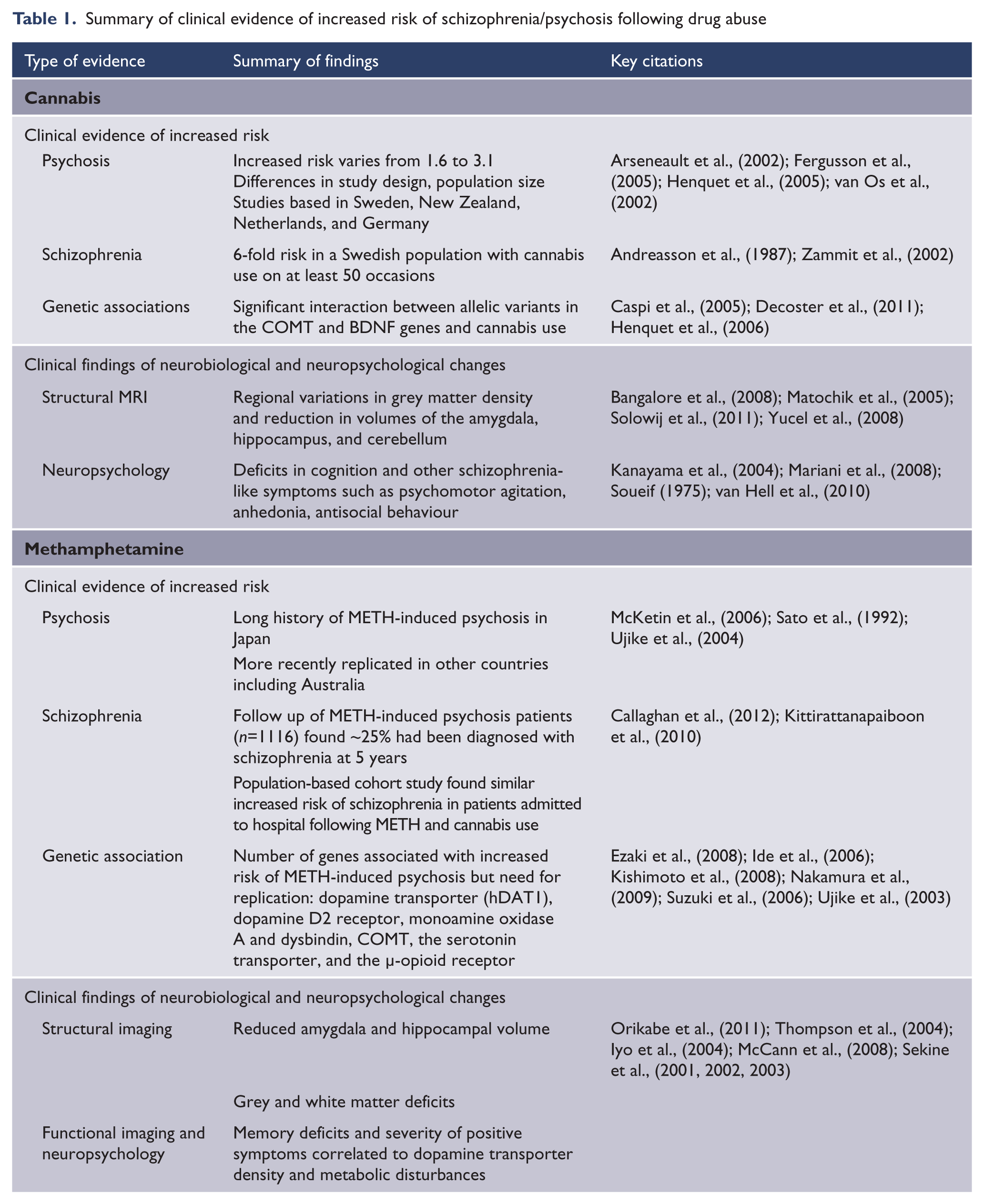
As summarised in Table 1, for nearly three decades, studies have investigated the causal link between the use of cannabis and schizophrenia development, with the one conducted by Andreasson et al., (1987) as the one most commonly cited. Involving more than 45,000 Swedish conscripts, aged 18–20, and with a follow up almost 15 years after the start of the study, this group found that the relative risk of developing schizophrenia increased with cannabis consumption. For example, the risk increased approximately 6-fold with use on more than 50 occasions. These findings were later confirmed by a follow-up study in which the subjects from the Andreasson study were analysed for a further 8 years (Zammit et al., 2002). In another prospective longitudinal study, adolescents who had begun using cannabis as early as 15 were more likely to develop schizophrenia in adulthood compared to controls (Arseneault et al., 2002). The strength of the association between cannabis use and psychotic symptoms was further assessed by Fergusson et al., (2005) in terms of the effects of other uncontrolled confounding factors as well as the direction of causality, i.e. whether cannabis use leads to the disorder or whether cannabis use is a consequence of the vulnerability to its effects as a result of the disorder. Using statistical techniques and mathematical modelling to account for various confounders, these authors reported that daily users of cannabis were almost twice as likely to have psychotic symptoms and that the risk was proportionate to consumption. The direction of causality was towards one where psychosis was a consequence of cannabis use.
Summary of clinical evidence of increased risk of schizophrenia/psychosis following drug abuse
Several studies have also suggested that cannabis use is associated with an earlier age of onset of schizophrenia symptoms (De Hert et al., 2011; Gonzalez-Pinto et al., 2008; Leeson et al., 2011; Sevy et al., 2010; Sugranyes et al., 2009; Veen et al., 2004) and several hypotheses have been suggested to account for this observation (Sevy et al., 2010; Veen et al., 2004). One suggests that it has almost nothing to do with cannabis use per se but rather the fact that its abuse is more prevalent and likely to become a lifelong habit in the younger demographic. Alternatively, it may be that the developing brain is much more susceptible to the harmful effects of cannabis (Ehrenreich et al., 1999), a theory which is consistent with the so-called ‘two hit’ neurodevelopmental hypothesis of schizophrenia (Bayer et al., 1999; Fatemi and Folsom, 2009; McGrath et al., 2003; Murray and Fearon, 1999). Specifically, in patients who have a genetic or environmentally induced predisposition for the disorder (‘first hit’), cannabis use (‘second hit’) may precipitate or accelerate the onset of schizophrenia (Figure 1) (Henquet et al., 2005, 2008; van Os et al., 2002).
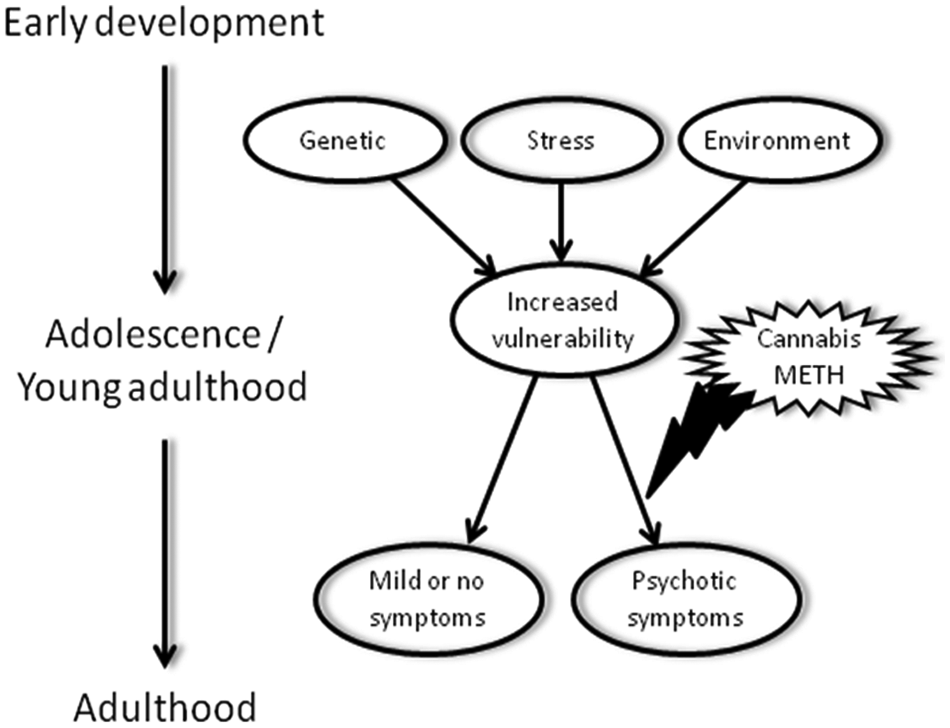
Hypothetical sequence of events illustrating how detrimental factors in early development, such as ‘risk’ gene predisposition, perinatal stress, or other environmental factors, may lead to increased vulnerability to the effects of abused cannabis or methamphetamine (METH), ultimately leading to psychotic symptoms/schizophrenia (bottom right). Genetic risk factors include polymorphisms in the COMT or BDNF genes. In the absence of drug abuse in adolescence or young adulthood, early developmental factors and their resulting increased vulnerability do not lead to symptom development (bottom left).
One such genetic predisposition is believed to involve polymorphisms of the gene for the enzyme, catechol-O-methyltransferase (COMT), which is involved in the metabolism of dopamine in the neuronal synapse. Substitution of the methionine residue at codon 158 for a valine residue (Val158Met) produces a variant of COMT high in enzymatic activity. Dysregulation of dopaminergic activity in the brain is a major pathological feature of schizophrenia (Howes and Kapur, 2009) and, in addition, can be induced by the psychoactive substituent of cannabis, delta-9-tetrahydrocannabinol (THC) (Kuepper et al., 2010). Caspi et al., (2005) aimed to determine whether there was a gene–environment interaction between the Val158Met polymorphism and cannabis abuse since the association between COMT polymorphisms and schizophrenia by itself was inconclusive (Sagud et al., 2010). The results showed that adolescent users of cannabis who carried the Val158 allele were approximately 10-times more likely to exhibit psychotic symptoms in adulthood. However, Henquet et al., (2006) suggested that while the risk still remained in carriers of the Val allele, it was restricted to carriers who had shown pre-existing psychosis liability based on the Community Assessment of Psychic Experiences (CAPE) psychometric scale. This led the authors to suggest the possible involvement of other genetic factors that moderate the THC–COMT interaction. However, in contrast to these two studies, one report showed that subjects with schizophrenia who were carriers of the low-activity variant of the COMT enzyme (Met-Met) were twice as likely to have used cannabis (Costas et al., 2011) and another study concluded there was no cannabis–COMT interaction (Kantrowitz et al., 2009).
More recently, the Val66Met polymorphism of brain-derived neurotrophic factor (BDNF) has been identified to interact with cannabis use and this effect was sex dependent (Decoster et al., 2011). In males, the age of onset was reduced as a result of cannabis use independent of BDNF status. However, in female users, the age of onset was significantly lower by almost 7 years in carriers of the Met allele BDNF polymorphism. BDNF has several roles, which include survival and differentiation of developing neurons as well as, importantly, regulation of synaptic plasticity in adult neurons (Favalli et al., 2012). Conceivably, polymorphisms would affect the functional activity of BDNF and this could underlie some of the pathological features of schizophrenia. In an attempt to explain the findings, Decoster et al., (2011) highlighted the role of BDNF and androgens in the hippocampus as well as the links between cannabis use and hippocampal morphology. They then extrapolated and hypothesised a possible three-way interaction between altered BDNF functionality, androgens, and cannabis use, which may translate into the earlier age of onset in females.
Despite all of the evidence that seems to suggest causation between cannabis use and schizophrenia, Cohen et al., (2008) in their review have highlighted an important discussion point: if increasing cannabis use really does cause schizophrenia, then population statistics should reveal this clearly. Degenhardt et al., (2003) in their analysis of Australian trends over a 30-year period found that the declining incidence of schizophrenia did not support such a causal link. However, Hickman et al., (2007) predicted, based on incidence and prevalence data of cannabis use (1970–2002) and schizophrenia (1997–1999) collected from three populations in England and Wales, that by 2010 the percentage of cases of schizophrenia directly attributable to cannabis consumption would be between 10–25%, depending on the extent of consumption. To date, there has been no follow-up study to determine the accuracy of this prediction.
Cannabis and schizophrenia neurobiology and neuropsychology
It is widely accepted that dysregulation of two main neurotransmitter systems in the brain – dopamine and glutamate – is associated with the pathophysiology of schizophrenia. The dopamine hypothesis proposes that positive symptoms of schizophrenia are mediated by subcortical hyperdopaminergia (Howes and Kapur, 2009). Several animal model studies have indicated direct and long-term effects of chronic cannabis abuse on dopaminergic activity in the brain. Interestingly, a case study reported increased dopaminergic neurotransmission in a patient with schizophrenia who had smoked cannabis during a pause in a SPECT imaging session; before and after images revealed an acute 20% decrease in striatal D2 receptor binding ratio, suggestive of increased dopamine release (Voruganti et al., 2001). However, this result was in contrast to [11C]-raclopride imaging studies that failed to find evidence of increased dopamine release in subjects with a history of recreational cannabis use or immediately after cannabis intake (Stokes et al., 2009, 2012).
There have been several structural MRI reports of cannabis-induced alterations in the morphology of several brain regions (summarised in Table 1). First-episode subjects with schizophrenia and recreational users with a history of cannabis use have been reported to display reductions in grey matter density in the right posterior cingulate cortex, right parahippocampal gyrus (Bangalore et al., 2008; Matochik et al., 2005) and increases in the precentral gyrus and thalamus (Matochik et al., 2005). Studies have also reported volume reductions in the amygdala, hippocampus, and cerebellum in chronic users with and without schizophrenia (Solowij et al., 2011; Yucel et al., 2008). In particular for those who started using cannabis earlier, brain volume and percentage cortical grey matter were lower whereas percentage white matter volume was higher (Cohen et al., 2012; Wilson et al., 2000). All of these brain regions contain varying densities of cannabinoid receptors (Glass et al., 1997). However, it should be noted that, in contrast to these findings, others have reported no effect on regional brain volume or density (Block et al., 2000; Tzilos et al., 2005).
Deficits in cognition are characteristic features of schizophrenia and there have been clinical studies that have shown that both early and chronic use of cannabis can have a significant impact on attentional processes (Emrich et al., 1997; Pope et al., 2001; Solowij et al., 2002). In particular, one imaging study reported that, in addition to increased activation of brain regions involved in working memory, chronic users of cannabis also activate brain regions unrelated to this task as a form of compensation (Kanayama et al., 2004). Studies have also reported other cannabis-related behaviours relevant to the schizophrenia phenotype such as psychomotor agitation, effects on visual-motor coordination (Soueif, 1975), anhedonia (van Hell et al., 2010), and antisocial behaviour (Mariani et al., 2008) (summarised in Table 1).
Methamphetamine and psychosis
As summarised in Table 1, much of the literature regarding methamphetamine (METH)-induced psychosis comes from Japan, due to the high levels of use following World War II and low rates of polydrug use (Sato et al., 1992; Ujike and Sato, 2004). However, since the late 1990s there has been an explosion of production and availability of amphetamine-type stimulants worldwide and this has coincided with increased METH use throughout Asia, Australia, and the USA (United Nations Office on Drugs and Crime, 2003). The number of reports of METH-induced psychosis in these emerging regions, including Australia, has increased over recent years (McKetin et al., 2006). For over four decades in Japan, and more recently around the world, clinicians have noted that while most METH users who suffer from psychosis will experience transient episodes that subside during drug abstinence, others will experience spontaneous relapse of symptoms in the absence of drug use and even persistent psychosis that continues for over a month following drug withdrawal. The more persistent psychosis has been described as closely resembling paranoid schizophrenia with comorbidity of negative and cognitive symptoms (Jacobs et al., 2008; Zweben et al., 2004). One study has found that almost 25% of METH users in Thailand who were hospitalised following an acute psychotic episode, had been diagnosed with schizophrenia at 5-year follow up (Kittirattanapaiboon et al., 2010). This is the strongest evidence to date that METH may trigger or increase the risk of schizophrenia. A more recent study reported that the risk of schizophrenia in METH users was equivalent to that of cannabis users compared to a proxy comparison group of appendicitis patients (Callaghan et al., 2012).
In Taiwan, it has been shown that earlier age of use, greater quantities of use, and familial risk for schizophrenia increase the likelihood of METH-users experiencing psychotic episodes (Chen et al., 2003, 2005). In one of these studies, of the 2000 METH users involved, 7% experienced persistent psychotic symptoms lasting more than 1 month after drug withdrawal, which was almost 20% of the users who had experienced any psychotic symptoms. Here, persistent psychosis was associated with a 2-times greater morbid family risk of schizophrenia compared to those whose symptoms were transient (Chen et al., 2005). These findings suggest a gene–environment interaction (Figure 1) and a number of genetic association studies have been performed to examine aspects of METH-abuse disorder, including psychosis. However, a major caveat of this work is that all studies have been conducted in Japanese populations and positive findings have not necessarily been replicated. In addition, many studies lacked sufficient power to identify positive associations (Bousman et al., 2009). Nevertheless, several genes have shown significant association with METH-induced psychosis, and a subset of these have specifically been associated with the development of persistent psychosis compared to the transient type, including variants of the genes for the dopamine transporter, dopamine D2 receptor, monoamine oxidase A, and dysbindin (Kishimoto et al., 2008; Nakamura et al., 2009; Ujike et al., 2003). In contrast, the development of psychotic symptoms with spontaneous relapse in the absence of METH use was associated with variants in the genes for COMT, the serotonin transporter, and the µ-opioid receptor (Ezaki et al., 2008; Ide et al., 2006; Suzuki et al., 2006). Interestingly, in the case of the COMT polymorphism, the methionine ‘low activity’ allele was associated with increased risk of developing METH-induced psychosis with spontaneous relapse, while there is conflicting evidence about which allele may confer risk of developing cannabis-induced schizophrenia psychosis. Together, these findings suggest that genetic risk factors affecting different signalling systems may contribute to the development of different types of psychosis following METH use. Specifically, it appears that alterations of dopamine signalling may confer risk to the development of persistent schizophrenia-like psychosis, consistent with the dopamine hypothesis of schizophrenia.
Methamphetamine and psychosis neurobiology and neuropsychology
Neuroimaging studies (summarised in Table 1) have shown that, in METH users, reductions in dopamine transporter binding levels in the frontal cortex and striatum negatively correlated with severity of positive symptoms (Sekine et al., 2001, 2003; Iyo et al., 2004). This parallels findings of reduced dopamine transporter binding in the striatum of schizophrenia patients (Laakso et al., 2001). Using magnetic resonance spectroscopy, further metabolic disruptions were detected in the basal ganglia that were found to correlate with severity of psychiatric symptoms in abstinent METH users (Sekine et al., 2002). Together, these findings suggest that subcortical pathology following METH use may contribute to the development of psychiatric symptoms. Pathology in the striatum may also contribute to cognitive deficits, with reduced striatal D2 receptor binding in this region found to correlate with reduced metabolism in the orbitofrontal cortex in abstinent METH users (Volkow et al., 2001) and reduced dopamine transporter binding correlating with memory deficits (McCann et al., 2008). One recent study has looked exclusively at METH-induced psychosis patients and observed bilateral reduction in hippocampal and amygdala volume (Orikabe et al., 2011), which again shows marked similarities to findings in schizophrenia (Yoshida et al., 2009). Others have shown extensive changes in grey and white matter in current METH users, including reduced hippocampal volume that correlated with memory deficits (Thompson et al., 2004). Future studies should focus on METH users who have experienced persistent psychosis and correlate neuroimaging changes to psychiatric rating to provide further insight into the neurobiology of METH-induced psychosis with relevance to schizophrenia. Recent studies have reported that there is considerable overlap in the symptomology of METH-induced psychosis and schizophrenia (Jacobs et al., 2008; Srisurapanont et al., 2011)
Summary
So far, the findings of several clinical studies have to varying degrees supported the existence of causation between cannabis and schizophrenia as well as METH use and psychosis. The variation is likely to be due to many factors, not limited to methodological differences in study design, as well cultural and demographic factors. With respect to cannabis, there are significant differences in methodology of the various studies especially in terms of the nature of cannabis use (e.g. amount smoked per day, duration). It is also important to note the regional differences in the potency of cannabis (Di Forti et al., 2009; ElSohly et al., 2000; Field and Arndt, 1980). Moreover, not all studies focused on patients with schizophrenia caused by cannabis abuse, but more generally on cannabis users, and future studies should shift more towards patients. It is quite clear that, compared to the investigative work that has been done for cannabis, evidence for METH-induced schizophrenia is more limited. This is likely due to the reduced prevalence of METH use compared to cannabis. However, this does not necessarily relegate its importance as a possible aetiological factor in the development of schizophrenia and psychosis, and further clinical and preclinical studies are certainly warranted. Indeed, additional evidence may begin to emerge, given the increasing prevalence of METH use worldwide during the last 20 years. In the future, clinical studies should aim to replicate some of the previous genetic association and neuroimaging findings from METH-induced psychosis, with a separation where possible of patients with persistent psychosis and spontaneous relapse compared to METH users who experience transient psychosis or no psychosis at all. In addition, long-term studies that track the METH users for a decade or more are necessary to collect sufficient evidence to understand the relationship between METH use and the development of schizophrenia and psychosis.
Animal models
Animal models are important for studying psychiatric diseases as they allow the use of methods that for ethical reasons cannot be used in humans and enable the researcher to examine hypotheses regarding the development of these diseases in a controlled environment that would not be possible in human studies. It is, however, critical to note that due to the complex human psychopathology of psychiatric diseases, it is not possible to model a disease like schizophrenia in its entirety. However, a number of methods can be used to create models for specific aspects and symptoms, such as selected cognitive deficits, psychomotor activity and anxiety, impaired prepulse inhibition, social withdrawal, and anhedonia, that have clinical relevance (Ellenbroek and Cools, 2000; Geyer and Moghaddam, 2002; van den Buuse, 2010; van den Buuse et al., 2005; also summarised in Table 2). We can also examine neuropathology in these models in an attempt to extrapolate and understand what might be occurring in humans. For the purpose of this review, our focus will be on animal models that have been created by either (i) repeated drug (cannabinoid or METH) administration or (ii) the modification of specific genes or an early development insult (‘first hit’) followed by repeated drug administration (‘second hit’).
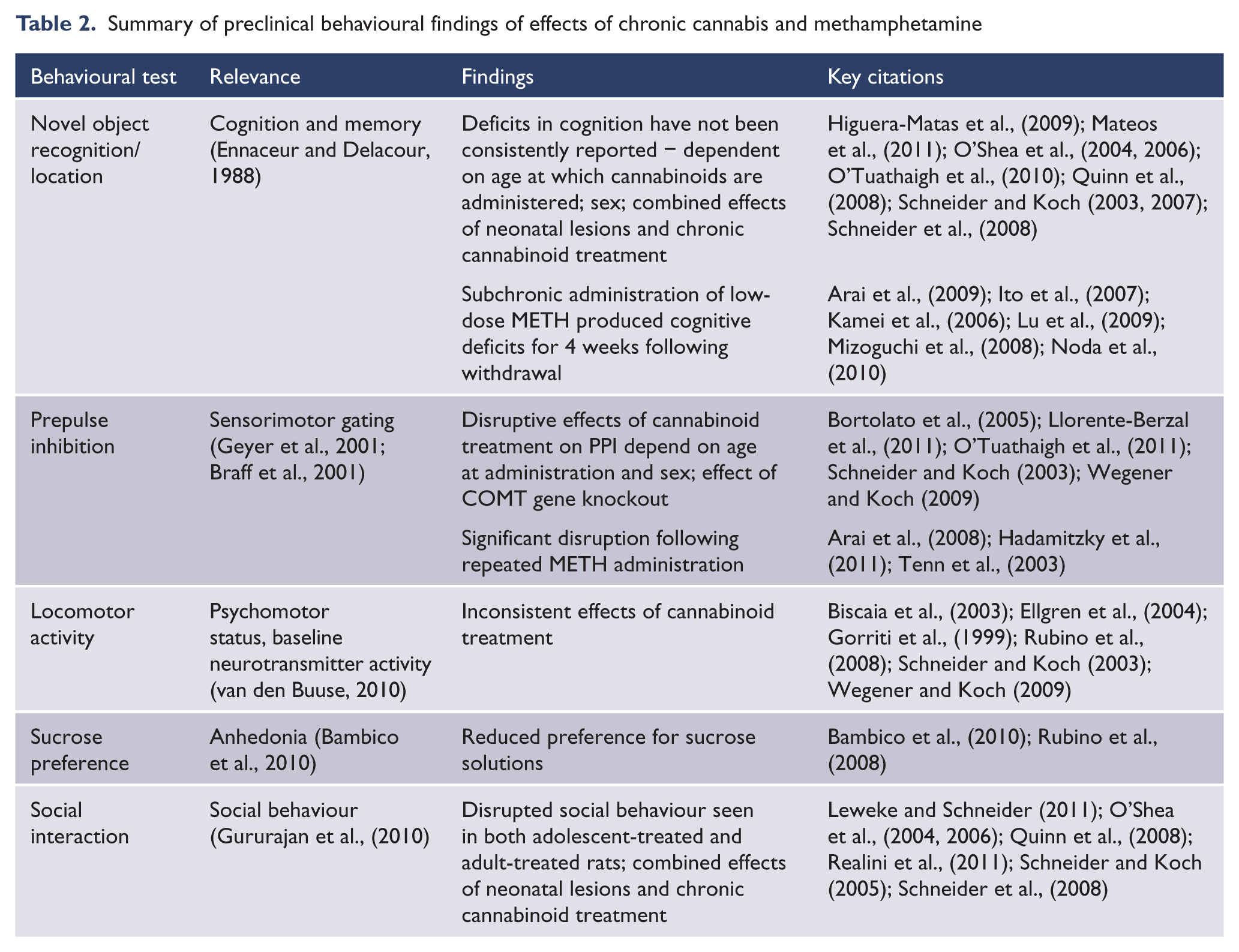
Summary of preclinical behavioural findings of effects of chronic cannabis and methamphetamine
Chronic cannabis and behaviour
Prior to discussing the findings of animal studies that have utilised chronic administration of cannabinoid compounds, it is important to note one important caveat – cannabis consumed by humans contains over 60 different phytocannabinoids (Ashton, 2001). However, in the majority of preclinical studies modelling the effects of cannabis in humans, synthetic specific agonists of cannabinoid receptors are used, such as CP55,940 and WIN55,212-2, as well as the archetypal cannabinoid THC in isolation. This could be seen as an oversimplification of a clinical effect and further complicated by differences in agonist potency and binding affinity for the cannabinoid receptors (Breivogel and Childers, 2000; Felder et al., 1995; Martin et al., 1991). However, this is the best initial approach in order to elucidate specific pharmacological mechanisms involved in cannabis. Once these have been well characterised across a range of methodologies, future studies could involve the evaluation of multiple cannabinoids in the same experiment.
Deficits in cognitive function in adulthood following chronic cannabinoid treatment during adolescence have been amongst the most consistently reported findings to support the clinical association that adolescent cannabis use confers significant risk for schizophrenia. Preclinically, cognition can be assessed in animals using several specific paradigms, such as the novel object recognition test (Ennaceur and Delacour, 1988) that is focused in particular on recognition memory. This two-trial test uses the innate tendency of rodents to spend more time with a novel object than with a familiar object. In rats, chronic pubertal exposure to the cannabinoid-receptor agonist WIN55,212-2 and THC induced significant impairment of recognition memory in the novel object recognition task in adulthood (Quinn et al., 2008; Schneider and Koch, 2003; Schneider et al., 2008). However, when the cannabinoid compound was chronically administered to adult rats there were no behavioural effects (Schneider and Koch, 2003). In contrast, O’Shea et al., (2006) found that chronic treatment with the cannabinoid-receptor agonist CP55,940 perinatally or during adolescence or early adulthood produced significant deficits in object recognition in male rats of all ages. This is in line with a study in mice performed by O’Tuathaigh et al., (2010) where chronic exposure to THC either during adolescence or adulthood resulted in impairments in the novel object task in male and female animals. Notably, this was one of the few studies where both sexes were analysed and compared with each other, while the other studies were in male animals only. It is important to examine both males and females as sex differences have been known to occur. For example, Mateos et al., (2011) found that only adult male rats exhibited impaired novel object recognition memory after CP55,940 treatment during adolescence while female rats showed impaired performance in another test, the object location task, in which males were unaffected. On the other hand, O’Shea et al., (2004) did find object recognition impairment in adult female rats after adolescent CP55,940 treatment. The only study that reported no effects of chronic adolescent cannabinoid treatment on recognition memory in adulthood in either sex was by Higuera-Matas et al., (2009). This study did, however, have the longest withdrawal period (59 days) between the last day of drug administration and behavioural testing and it is possible that cognitive deficits could have been detected at an earlier time point similar to the other studies. The question therefore arises as to whether the cognitive deficits found in most studies are irreversible or whether they can recover after a prolonged period of time.
Prepulse inhibition (PPI), which can be measured in humans as well as in animals, is the phenomenon of a reduction in startle response when a loud stimulus is preceded by a stimulus of lesser amplitude (prepulse) (Braff et al., 2001). PPI is considered a pre-attentive measure of sensorimotor gating, a cognitive filtering mechanism that has been shown to be disrupted in patients with schizophrenia (Braff et al., 2001). In male rats, pubertal treatment with WIN55,212-2 resulted in disrupted PPI in adulthood that could be reversed by acute treatment with the antipsychotic haloperidol (Schneider and Koch, 2003; Wegener and Koch, 2009). In adult rats, WIN55,212-2 treatment had no effect on PPI (Bortolato et al., 2005). This result in particular reinforces the proposition of an age-dependent vulnerability to the effects of cannabinoids. In the only study measuring PPI in both male and female rats, cannabinoid exposure during adolescence reduced PPI in female rats but not males (Llorente-Berzal et al., 2011).
In terms of the positive symptoms, locomotor hyperactivity seen in animals at baseline or following treatment with psychostimulants has been used both as a model of psychomotor agitation and as a bio-assay of disrupted dopaminergic and glutamatergic activity. This is based on the assumption that enhanced subcortical dopaminergic activity or antagonism of glutamatergic receptors of the NMDA class, leads to behavioural hyperactivity (Beninger, 1983; Ellenbroek and Cools, 2000; van den Buuse, 2010). Only a few papers have investigated the long-term effects of chronic cannabinoid treatment on locomotor hyperactivity and the reports are not conclusive. While some showed that adolescent exposure to cannabinoid-receptor agonists induced no effects on locomotor activity (Biscaia et al., 2003; Rubino et al., 2008; Schneider and Koch, 2003), others found that chronic pubertal cannabinoid treatment induced locomotor hyperactivity in adulthood at baseline (Wegener and Koch, 2009). In addition, enhanced responses to an acute challenge dose of amphetamine on locomotor activity and stereotyped behaviour were observed in adult animals pre-treated with THC (Gorriti et al., 1999), although this finding was not replicated by others (Ellgren et al., 2004).
Adolescent treatment with cannabinoids in animals can also induce behavioural deficits with relevance to some of the negative symptoms seen in schizophrenia. For example, chronic cannabinoid treatment during adolescence has been shown to induce a state of anhedonia-like behaviour, i.e. an inability to experience pleasure. In animal models, this can be assessed using a sucrose preference test and it was shown that, when treated with THC, male and female rats showed reduced preference for a sucrose solution (Bambico et al., 2010; Rubino et al., 2008). Another key negative symptom is social withdrawal, which can be measured in animals using social interaction tests (Geyer and Moghaddam, 2002; Gururajan et al., 2010; van den Buuse, 2010). Reduced social interaction has been observed in adult rats when they were exposed to cannabinoids in adolescence and this was shown in both male and female rats (Leweke and Schneider, 2011; O’Shea et al., 2004, 2006; Realini et al., 2011; Schneider et al., 2008). Studies on the same treatment in adult rats have produced conflicting results. While O’Shea et al., (2006) and Quinn et al., (2008) showed that social interaction was impaired in male rats after adolescent as well as after adult exposure to CP55,940 and THC, respectively, Schneider et al., (2008) only observed reduced social interaction after adolescent, but not adult, treatment with WIN55,212-2.
Overall, the available behavioural data support the hypothesis that adolescent exposure to cannabinoids can lead to long-term behavioural effects in adulthood and could be a risk factor for developing schizophrenia-like symptoms. Even if in general terms cannabis abuse cannot be said to directly cause schizophrenia, it might be able to trigger the onset of the disease in a predisposed minority of people. This ‘two hit’ hypothesis (Figure 1) suggests that adverse early environmental or genetic factors cause the development of abnormal neural networks, thus leaving the brain more vulnerable towards a ‘second hit’ later on in life, which could then lead to the onset of schizophrenia (Bayer et al., 1999; Murray and Fearon, 1999). Only a few animal studies have addressed this in regards to cannabinoid treatment as a ‘second hit’. Schneider and Koch developed a model of this hypothesis and analysed the effects of neonatal prefrontal cortex lesions combined with chronic cannabinoid treatment during puberty (Schneider and Koch, 2005, 2007). They could show that the combined treatments had detrimental effects on various forms of social behaviours (Schneider and Koch, 2005) and novel object recognition memory (Schneider and Koch, 2007).
We recently explored the ‘two hit’ interaction of neonatal environment and young-adult treatment with the cannabinoid-receptor agonist CP55,940 on adult locomotor and exploratory behaviour (Figure 2) (Klug and van den Buuse, 2012). Chronic cannabinoid receptor stimulation in young adulthood induced a tendency for the adult animals to explore a new environment less. There were qualitative differences between male and female rats, with the former showing reduced ambulatory movements but the latter showing reduced exploratory rearing. Significantly, none of the behavioural changes were influenced by neonatal maternal deprivation during the first 2 weeks of life, arguing against a ‘two hit’ interaction for this behaviour. It should be noted that the reduced exploratory activity in both male and female rats could reflect enhanced anxiety levels rather than be indicative of cognitive or psychosis-like changes. Therefore, further experiments should address other behavioural paradigms, including psychotropic drug-induced locomotor hyperactivity as a model of psychosis.
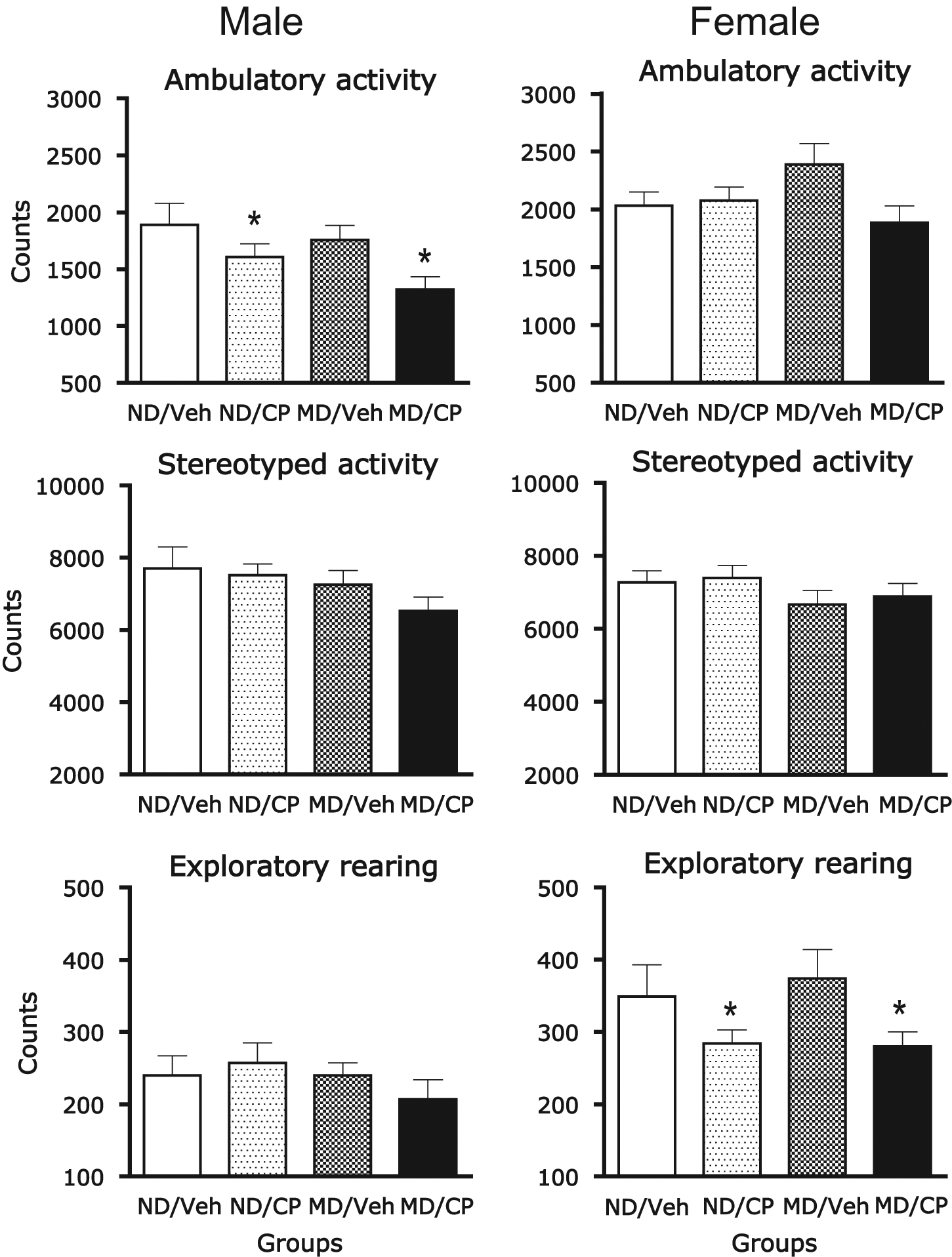
Adult rats show behavioural changes after adolescent/young adult cannabinoid treatment
When genetic factors were considered, mice with a knockout of the COMT gene were shown to be more vulnerable than wild-type control mice towards adolescent cannabinoid treatment and displayed disrupted PPI (O’Tuathaigh et al., 2011) and disrupted spatial working memory (O’Tuathaigh et al., 2010) in adulthood. These studies highlight the interactions between cannabis exposure and genetic background even though the complete knockout of the gene does not mimic the human situation, where either a polymorphism for high-activity COMT variants (Caspi et al., 2005) or the low-activity variants (Thompson et al., 2004) are implicated in cannabis abuse and schizophrenia. Another gene implicated in the development of schizophrenia is neuregulin 1 (Nrg1) (Solowij et al., 2011) and acute treatment with cannabinoid-receptor agonists induced an enhancement in PPI in Nrg1 heterozygous mice compared to wild-type controls (Boucher et al., 2007, 2011) while repeated cannabinoid exposure abolished these effects (Boucher et al., 2011). In a more recent study, Long et al., (2012) investigated the effects of chronic cannabinoid exposure during adolescence in Nrg1 mutant mice and found no effects on PPI. In contrast to their theory that mutant mice would be more susceptible to the effects of THC, these authors found that THC-induced reduction in social behaviour was in fact ameliorated in mutant mice.
Chronic cannabis neurobiology
As described above, adolescent cannabis use may lead to deficits in cognition or the emotional domain. These deficits may be induced by effects in several brain regions, changes in synaptic plasticity and/or interactions with neurotransmitter systems such as γ-aminobutyric acid (GABA), glutamate, and dopamine. For example, memory impairment after adolescent exposure to THC was accompanied by lasting changes in the hippocampal protein expression profile related to oxidative stress (Quinn et al., 2008) as well as reduced spine density and reduced numbers of dendrites in the hippocampal dentate gyrus, suggesting less effective synaptic connectivity (Rubino et al., 2009). In addition, these animals also showed a decrease in NMDA receptor levels together with reduced postsynaptic protein expression, suggesting a decrease in NMDA receptor function (Fan et al., 2010; Rubino et al., 2009) consistent with the glutamatergic hypofunction hypothesis of schizophrenia (Newcomer and Krystal, 2001; Stahl, 2007). As the NMDA receptor plays a pivotal role in learning and memory, these findings might also explain the observed memory impairments (Newcomer and Krystal, 2001; Stahl, 2007). In a more recent study, previous adolescent exposure to THC reduced NMDA receptor density only in the hippocampus of female, but not male, adult rats and this was accompanied by cognitive impairments in female, but not male, rats (Zamberletti et al., 2011). Others have found similar results investigating glutamatergic and GABAergic systems in the hippocampus of adult rats treated with a cannabinoid-receptor agonist during adolescence (Higuera-Matas et al., 2012). Here, NMDA receptor downregulation was also exclusively found in female animals. These cannabinoid-treated females showed increased GABA receptor density as well as increased GABA and decreased glutamate release, suggesting a glutamate/GABA imbalance in female rats that could be responsible for some of the behavioural abnormalities observed after adolescent cannabis abuse (Higuera-Matas et al., 2012). When looking at GABAA receptor density only 24 hours after the last injection of a chronic cannabinoid regime treatment, adult rats displayed increased GABAA receptor levels in the hippocampus while adolescent rats did not (Verdurand et al., 2010). Thus, it seems that while adolescent cannabis use might have no short- term effects on GABA receptors it can produce long-term alterations that only emerge in adulthood.
The exposure to exogenous cannabinoids might also cause effects on the dopaminergic system. In rats, acute administration of cannabinoid-receptor agonists has been shown to increase dopamine release in several brain regions, including the striatum and prefrontal cortex (Cheer et al., 2004; Pistis et al., 2002; Szabo et al., 2002; Tanda et al., 1997). In contrast to these acute effects, chronic treatment with cannabinoid-receptor agonists induced a significant reduction of dopamine metabolism in the prefrontal cortex but not in striatal regions (Jentsch et al., 1998; Verrico et al., 2003). As mentioned, a prefrontal hypodopaminergic state has been proposed as one on the neurobiological features of schizophrenia (Abi-Dargham and Moore, 2003). In regards to the impact of cannabinoid exposure on dopamine receptor levels, the effects seem to be sex- and region-specific as well as dependent on the treatment regime used. When receptor levels were measured after a short withdrawal period of 24 hours after chronic cannabinoid exposure, adolescent animals showed no changes in either D1 or D2 receptor levels while treated adult animals showed increased D1 and D2 binding (Dalton and Zavitsanou, 2010). Two studies have investigated the more long-term effects of early cannabinoid treatment and looked at receptor binding in adult animals after a long withdrawal period. While pre-adolescent exposure to cannabinoids (from postnatal day 28–38) increased D1 receptor levels in the nucleus accumbens shell of male adult animals and decreased D2 receptor levels in the CA1 region of the hippocampus of both sexes (Higuera-Matas et al., 2010), adolescent treatment (from postnatal day 37–47) increased D1 receptor levels in female adult animals and also increased D2 receptor levels in the prefrontal cortex of male adult rats (Zamberletti et al., 2011). Once again, these studies suggest that changes in the brain only emerge in adulthood after previous adolescent cannabis use while they seem to have no direct short-term effects during adolescence.
Chronic methamphetamine and behaviour
Similar to studies of the effects of cannabinoids in rodents, the most robust findings following METH treatment are the development of cognitive deficits. In adult mice, 1 week of daily low-dose METH administration was found to produce robust deficits in the novel object recognition task, which lasted at least 4 weeks following drug withdrawal (Arai et al., 2009; Ito et al., 2007; Kamei et al., 2006; Lu et al., 2009; Mizoguchi et al., 2008; Noda et al., 2010), and similar findings have also been reported in rats (Belcher et al., 2006). Investigation of the mechanisms underlying novel object recognition deficits in METH-treated mice revealed that these deficits were due to reduced task-dependent activation of the signalling factor ERK1/2 in the frontal cortex, and that this activation may be dependent on D1 receptor activation (Kamei et al., 2006). GABAB receptor signalling deficits have also been implicated in deficits in this model (Arai et al., 2009). These findings suggest that therapeutics that enhance ERK1/2 activation in the frontal cortex, such as galantamine and minocycline, may be useful in the treatment of cognitive deficits in schizophrenia and METH-abuse disorder (Mizoguchi et al., 2008; Noda et al., 2010).
Other aspects of learning and memory have been examined in rats, with repeated METH treatment producing deficits in working memory in the radial arm maze (Mizoguchi et al., 2011; Nagai et al., 2007) and METH self-administration causing a disruption in object location recognition (Reichel et al., 2012). Working memory deficits were rescued by co-administration of nicotine and clozapine but not haloperidol, suggesting that this model of cognitive deficits may also be useful for screening of precognitive drugs from the treatment of schizophrenia (Mizoguchi et al., 2011; Nagai et al., 2007). These findings also give the model some predictive validity, given previous reports that clozapine has superior efficacy in the treatment of cognitive deficits compared to other current antipsychotics (Meltzer and McGurk, 1999).
The effect of repeated treatment with METH on PPI has been examined following a number of different treatment regimes. One group has looked at the effect of a short repeated dosing regime of METH on baseline PPI in rats at 7 days withdrawal and found a modest disruption (Abekawa et al., 2008; Tomohiro et al., 2011). These deficits could be rescued by co-administration of various typical and atypical antipsychotic drugs, giving the model predictive validity. Others have looked at the effects of repeated METH administration immediately following the final drug exposure and have found more significant disruption of PPI (Arai et al., 2008; Hadamitzky et al., 2011; Tenn et al., 2003), whereas one study reported the disruption persisted 60 days following withdrawal (Tenn et al., 2005).
Chronic methamphetamine neurobiology
Treatment with amphetamines induces increased synaptic monoamine levels by acting on the vesicular and synaptic monoamine transporters.
Some groups have looked at the effects of METH in genetic mutant strains with relevance to genetic association findings in METH users. For example, the effects of acute and repeated METH on locomotor activity were attenuated in dopamine transporter heterozygous mutant mice (Fukushima et al., 2007). D2 receptor knockout mice also showed an attenuation in the acute response to METH; however, these mice showed an increase in sensitisation compared to WT animals, when animals receiving their first exposure to METH were compared to animals that had been pretreated (Kelly et al., 2008). This suggests a dissociation in the role of the D2 receptor between the acute and long term effects of METH. µ-Opioid receptor knockout mice also showed an attenuated response to acute METH and did not sensitise following repeated exposure, and in both these circumstances there appeared to be a ceiling effect on the hyperactivity that could be induced by METH (Shen et al., 2010). However, this reduced sensitivity to METH was also apparent in a reduced induction of stereotyped behaviours by high doses of METH. Unfortunately other behaviours have not been examined in these animals, and it should be a priority of future studies to look at other behavioural endophenotypes in METH-treated genetic mutant mouse strains.
Conclusion
Schizophrenia is a uniquely complex disorder and here we have reviewed evidence from both the clinical and preclinical literature that implicates the abuse of drugs such as cannabis and methamphetamine as a potential causative factor. Progress has been made with our understanding of the neurobiological mechanisms underlying the effects of long-term cannabis and methamphetamine use on psychosis development. This has been possible in part by basic science studies using animal models based on clinical observations of drug-induced psychosis and schizophrenia. The evidence suggests that there is a considerable role of age, sex, environmental influences, and genetics, and these are factors that deserve further investigation. Amongst several further directions of research, more genetically modified animal models can be used to investigate gene–drug interactions, given the significant role of genetic predisposition (Caspi et al., 2005; Decoster et al., 2011; Henquet et al., 2006; Kishimoto et al., 2008; Nakamura et al., 2009; Ujike et al., 2003). We still are largely in the dark about what biological mechanisms lie behind drug-induced psychosis and schizophrenia, and studies that look at long-term neuromolecular changes as well as imaging studies in these animal models is a way forward. We could then begin to, with caution, extrapolate the findings to a human scale and, potentially, assist in guiding treatment or public health policy by identifying at-risk individuals early on in life and ensuring that they are adequately informed of the risks. An on-going dialogue between basic and clinical researchers may help to identify at-risk individuals and novel pathways for treatment and prevention.
Footnotes
Funding
Parts of the work in this paper were supported by a project grant from the NHMRC (grant number 566879). MvdB is a Senior Research Fellow of the National Health and Medical Research Council of Australia (NHMRC). EM is recipient of an Australian Postgraduate Award. MK is a recipient of a Swinburne University of Technology international scholarship. The Mental Health Research Institute receives Operational Infrastructure Support from the Victorian State Government.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.