
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare but fatal disorder characterized by the proliferation and infiltration of macrophages and hyperactivated T lymphocytes that escape from the physiological control pathways and favour the existence of an environment of excessive inflammation and tissue destruction. HLH has been classified into two types: a primary or familial autosomal recessive form, caused by mutations in genes encoding proteins involved in the granule-dependent cytotoxic pathway (familial hemophagocytic lymphohistiocytosis [FHL] types 1–5); and other secondary or acquired form, generally associated with infections, malignancy, autoimmune diseases, metabolic disorders or primary immunodeficiencies. Since the first familial hemophagocytic lymphohistiocytosis-2 (FHL2) causative mutation in the PRF1 gene was described in 1999, more than 200 mutations have been identified to date. Here, we report the first case of very late-onset FHL2 in a Spanish 72-year-old female with splenomegaly, hypertriglyceridemia, hypofibrinogenemia, pancytopenia and marrow hemophagocytosis harbouring in heterozygosity two PRF1 variants proposed as causative in this study. The heterozygous mutation c.445G>A (p.Gly149Ser) identified in the exon 2 results in a missense mutation previously described as a probable pathogenic variant associated with the development of FHL2. Affecting the same exon, c.272C>T (p.Ala91Val) is the most prevalent variant of this gene. Although it was initially classified as benign, recent studies support its potential pathogenic role, considering it a variant of uncertain significance associated with a risk of developing FHL2. The genetic confirmation of FHL made possible an adequate counselling to the patient and direct relatives and provided important information for her control and follow-up.
Background
Hemophagocytic lymphohistiocytosis (HLH; OMIM#603553) is a rare but fatal disorder characterized by the proliferation and infiltration of macrophages and hyperactivated T lymphocytes that escape from the physiological control pathways and favour the existence of an environment of excessive inflammation and tissue destruction. 1 Clinically, HLH presents with prolonged fever, hepatosplenomegaly and cytopenias. Laboratory analyses generally show hyperferritinemia, hypertriglyceridemia, elevated levels of cytokines and hypofibrinogenemia. Appearing at any age, it typically starts before the first year of life. Initial therapy usually focuses on suppressing the overactivation of the immune system by combining chemotherapy and immunotherapy (etoposide, corticosteroids, cyclosporine and intrathecal methotrexate in patients with neurological involvement), but allogeneic hematopoietic stem cell transplantation (HSCT) is considered the only curative treatment available, which should be carried out as soon as possible due to the high mortality of patients with HLH due to progressive multiorgan failure.2,3
Hemophagocytic lymphohistiocytosis has been classified into two types: a primary or familial autosomal recessive form (FHL), caused by mutations in genes encoding different proteins that participate in granule-dependent cytotoxic function of T lymphocytes (CTLs) and natural killer (NK) cells; and other secondary or acquired form, generally associated with infections, malignancy, autoimmune diseases, metabolic disorders or primary immunodeficiencies. However, episodes of FHL are frequently triggered by infections, making it difficult to differentiate both pathologies in clinical practice since there are no discriminatory clinical or histological findings. 1 Up to a quarter of HLH cases are estimated to be familial, with varying frequencies of each subtype between different populations. 3 Depending on the affected gene, five subtypes of familial hemophagocytic lymphohistiocytosis (FHL) (FHL1–5) have been described to date: PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), FHL1 has been mapped to chromosome 9q and STXBP2 (FHL5). The gene responsible for FHL1 has not yet been identified, but it is believed to be linked to the 9q21.3-22 region. 4
Since Stepp et al. described the first familial hemophagocytic lymphohistiocytosis-2 (FHL2) causative mutation in the PRF1 gene in 1999, 5 more than 200 mutations have been identified. 6 The perforin gene has three exons, being only two and three translated into a 555-amino acid mature protein with three domains. The N-terminal cholesterol-dependent cytolysin domain, also called membrane attack complex-like domain (MACPF/CDC), allows the polymerization of perforin in the target cell membranes and facilitates the entry to the cytosol of granzymes, granulolysins and other lytic molecules produced by CD8 T lymphocytes and NK cells into the cytosol to induce apoptosis, in a mechanism known as granule-dependent cytotoxicity.6,7 This is followed by the epidermal growth factor (EGF)-like and the calcium-dependent C-terminal (C2) domains. The latter transiently binds the membrane lipids in the presence of calcium and promotes the self-assembly of perforin into cylindrical pores of 20 nm. 8 In addition to allowing the entry of lytic molecules, the pore formation favours an ionic exchange that results in an osmotic imbalance promoting cell destruction. Thus, the presence of perforin seems to be essential for granzymes and other lytic molecules to induce apoptosis in target cells. 6
In this study, we report the first case of very late-onset FHL2 in a Spanish 72-year-old female with splenomegaly, hypertriglyceridemia, hypofibrinogenemia, pancytopenia and marrow hemophagocytosis. The genetic study revealed the aetiology of its clinical context with the finding of two variants in the PRF1 gene. The genetic confirmation of FHL made possible an adequate counselling to the patient and her direct relatives, and provided important information for her control and follow-up.
Materials and methods
Subject
This study was performed in a woman who fulfilled the diagnostic criteria of HLH according to the Histiocyte Society’s HLH-949 9 and HLH-20041 10 protocols and her biological son. Written informed consent was obtained from both. Genetic studies could not be performed in the rest of the patient’s first-degree relatives.
A 72-year-old female subjected to radical cystectomy and urinary diversion for invasive bladder carcinoma 6 years before was hospitalized with persistent fever (8 days) and condition deteriorated, despite having started antibiotic treatment 5 days before. The patient was being studied for mild pancytopenia and recurrent panniculitis (nodular vasculitis) and had received various treatments: corticosteroids, immunomodulators and tuberculosis chemoprophylaxis. The bone marrow study performed 4 years earlier revealed mild myelodysplastic changes.
During hospitalization, the patient initially improved but in the following days, persistent fever, asthenia, weight loss, headache, liver dysfunction, general discomfort and confusional syndrome were observed. Serological test for herpes simplex virus types 1 and 2, cytomegalovirus, Epstein–Barr virus (EBV), varicella-zoster virus and human herpesvirus 6 showed past infection.
Laboratory test results showing the partial response achieved during the first episode follow-up period before and after starting treatment with dexamethasone and etoposide.
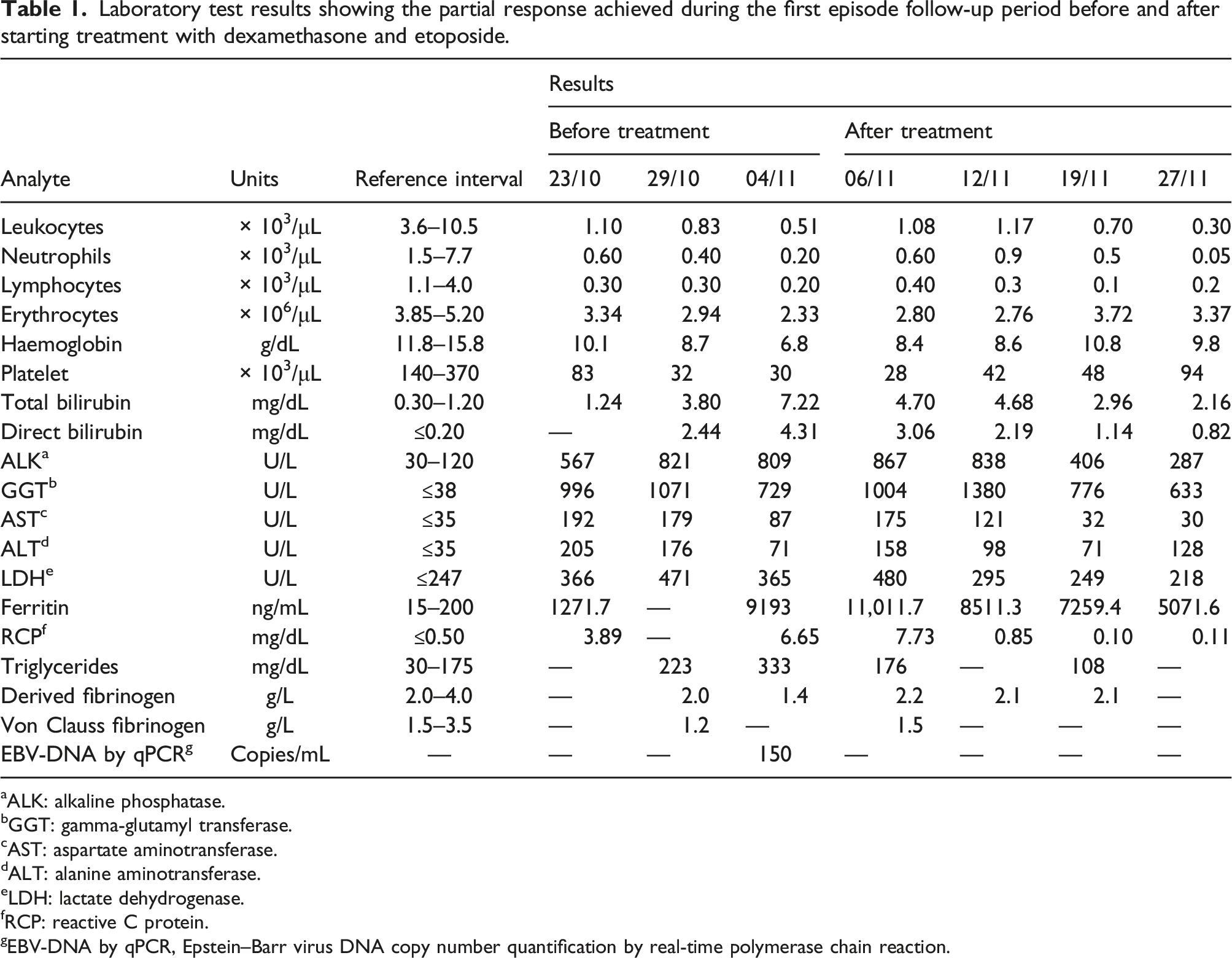
aALK: alkaline phosphatase.
bGGT: gamma-glutamyl transferase.
cAST: aspartate aminotransferase.
dALT: alanine aminotransferase.
eLDH: lactate dehydrogenase.
fRCP: reactive C protein.
gEBV-DNA by qPCR, Epstein–Barr virus DNA copy number quantification by real-time polymerase chain reaction.
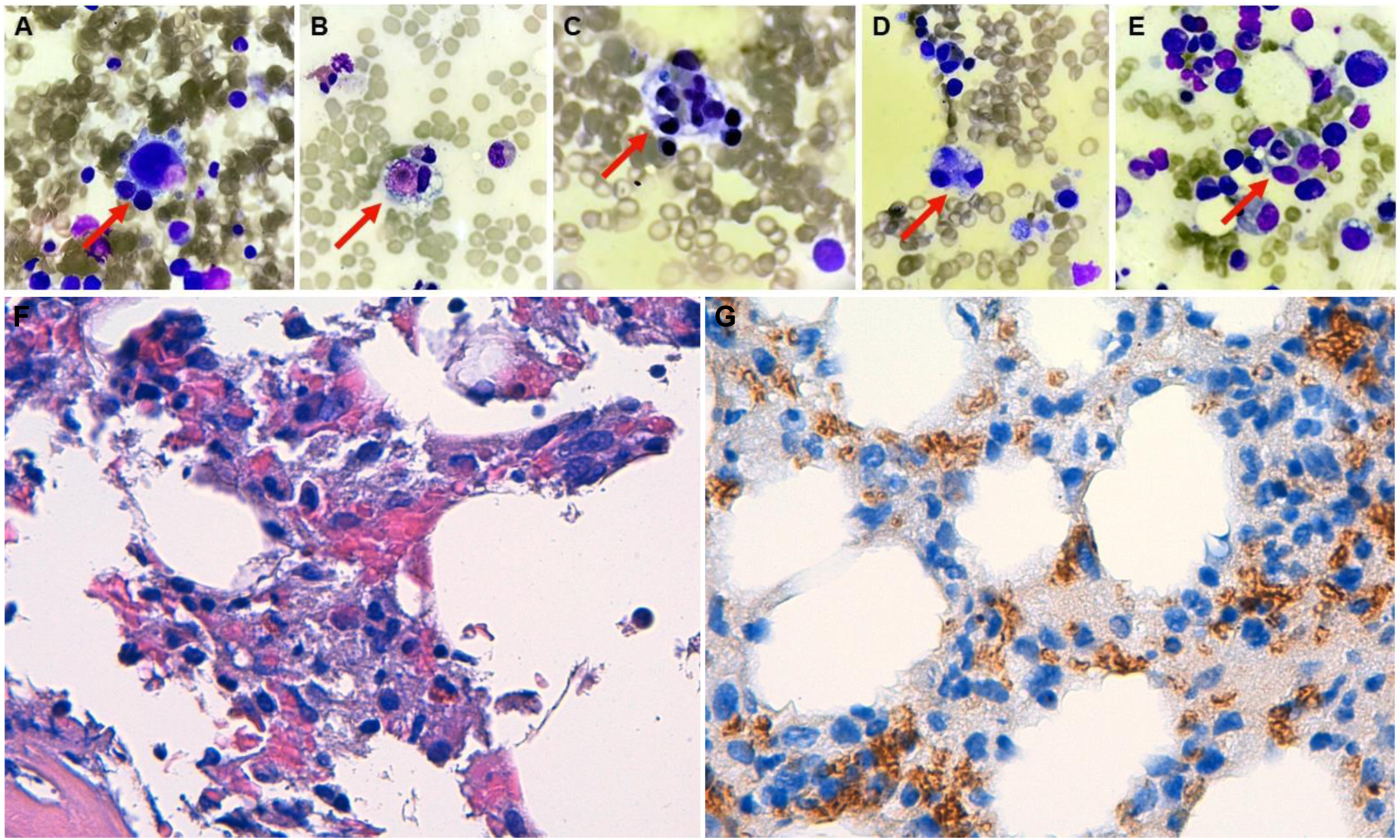
(a)–(e) Hemophagocytosis in the hypercellular bone marrow aspirate of the patient. Wright staining of bone marrow cells, with red arrows indicating dense histiocytes which have phagocytosed leukocytes and erythrocytes (magnification power: 1000×). (f) Hypercellular bone marrow histology with increased histiocytic cellularity and presence of hemophagocytosis (hematoxylin-eosin, 400X). (g) Glycophorin A (CD235a) immunostain in a similar field, with evidence of hemophagocytosis (glycophorin A, 400X).
Biochemical and haematological analysis
Before and after therapy, complete blood count was performed using UniCel® DxH 800 Coulter® analyser (Beckman Coulter Inc., USA). Absolute and relative numbers of blood cells were calculated from EDTA samples on the basis of total and differential counts. For biochemical analyses including total bilirubin, direct bilirubin, alkaline phosphatase, gamma-glutamyl transferase, aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, ferritin, reactive C protein, triglycerides, serum immunoglobulins and others, AU5820 and DxI800 analysers (Beckman Coulter Inc., USA) were used.
Molecular genetic analysis
Genomic DNA was isolated from blood samples using the Maxwell® RSC Blood DNA Kit and Maxwell® 16 System (Promega, USA). Fluorometric quantification of the DNA was determined by Qubit Fluorometric Quantitation using the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, USA). The entire coding region and adjacent intronic regions (5bp) of 34 genes related with FHL (ADA, AP3B1, AP3D1, BLOC1S6, BTK, CD27, CORO1A, FADD, FAS, FASLG, GATA2, IL2RA, IL2RG, ITK, LIPA, LYST, MAGT1, MEFV, MVK, MYO5A, NLRP3, PNP, PRF1, RAB27A, RECQL4, SH2D1A, SLC7A7, STX11, STXBP2, TBXAS1, TNFRSF1A, UNC13D, WAS, XIAP) were analysed by massive sequencing using the Illumina NextSeq® 550 platform and the SOPHiA™ clinical exome panel CES_v2 (LabGenetics, Spain). All potentially pathogenic single nucleotide variants were validated by Sanger sequencing. Analysis and interpretation of the found variants were conducted using the SOPHiA DDM software, ACMG score, Alamut® Visual 2.10, HGMD® Professional 2017.2, gnomAD browser and ClinVar.
Bioinformatic tools and structural biology analysis were used to hypothesize about the structural and functional consequences of the previously reported PRF1 variants found in the proband. Different in silico prediction tools were used including MutationTaster, PolyPhen-2, FATHMM, Mutation Assessor, SIFT, PROVEAN, Align-GVGD and others. In addition, to establish the degree of interspecies conservation of the variants, sequence alignments were performed comparing nine reported amino acid sequences for the perforin MACPF domain from different species (UniProtKB database). Finally, we collected information of PRF1 gene, protein sequence, domains, comparative modelling of 3D protein structure and additional annotations reported in UniProtKB and SWISS-MODEL Repository.
Results
Because patient’s condition was critical, empirical treatment with dexamethasone and etoposide was started following HLH-94 protocol. 9 Chemo-immunotherapy was also administered for 8 weeks. Anaemia, splenomegaly and hyperferritinemia persisted after initial therapy (partial response achieved). Although the patient was not candidate for HSCT, continuation therapy (HLH-04 protocol) 10 was started with etoposide and dexamethasone for 3 days (every 2 weeks) plus daily cyclosporine. Two months later, disease progression with central nervous system involvement was triggered by EBV reactivation. Quantification of blood EBV DNA was performed by real-time PCR. The results showed an increased from initial 150 log copies/microgram of DNA at the debut to 120,000 at this point.
Ganciclovir and intrathecal methotrexate plus weekly cytarabine were administered. Finally, the patient suffered general progressive damage and died few months after.
Molecular genetic analysis revealed the presence, in heterozygosity, of two variants that could justify the clinical picture of the patient. The heterozygous missense variant c.445G>A (p.Gly149Ser) identified in exon 2 of PRF1 gene (NM_001083116.2) has been described as a probable pathogenic variant associated with the development of FHL2 in ClinVar (RCV000622519.1 and RCV000819599.1) and Human Gene Mutation databases (HGMD; CM023668). 11 Affecting the same exon, c.272C>T (p.Ala91Val) is the most prevalent variant found in this gene, with a frequency population of 4–17%. Although initially classified as a benign variant, recent studies support its potential pathogenic role when found in compound heterozygosity with another variant in PRF1 gene, since it leads to a reduction of up to 50% in perforin activity.7,12,13 In ClinVar (RCV000014719.5) and HGMD (CM022053) databases, this variant is considered of uncertain significance, associated with a risk of developing FHL2.
Both variants were also designed as pathogenic by most of the in silico predictors used to hypothesize about the potential effect of the variants in the structural, theoretical and conformational parameters of the perforin (Figure 2). The phylogenetic analysis revealed a high degree of conservation of Ala91 and Gly149 in the MACPF/CDC domain, supporting the evolutionary and structural importance of these amino acids for the perforin polymerization and function. Conservation (a) and functional impact of the c.272C>T (p.Ala91Val) and c.445G>A (p.Gly149Ser) variants evaluated using different in silico prediction tools (b). *For Align GVGD, class C0 indicates that a change is unlikely to be pathogenic, class C65 represents the highest likelihood of a change to be pathogenic and class C55 indicates a moderate-high likelihood for the change to be pathogenic.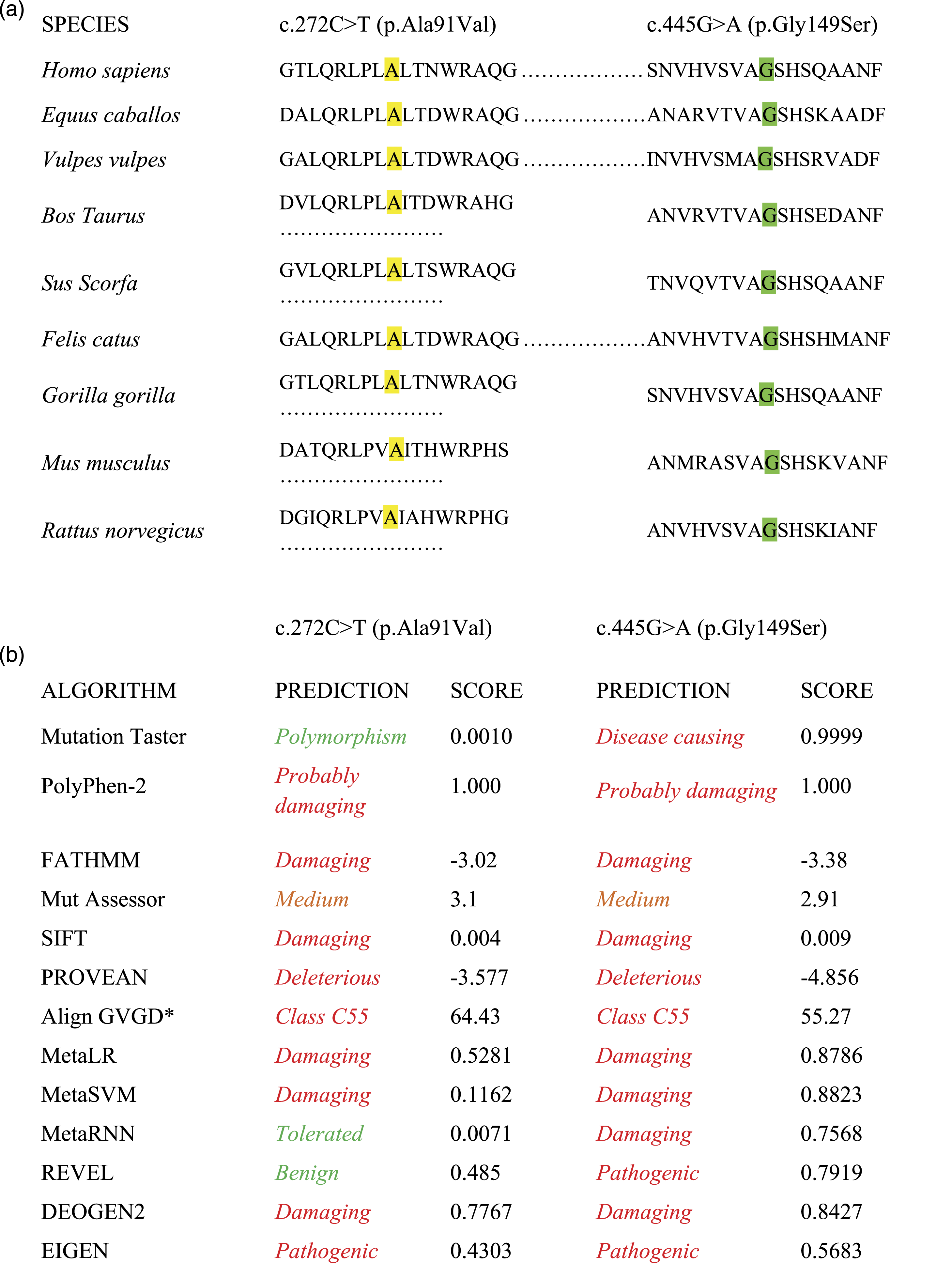
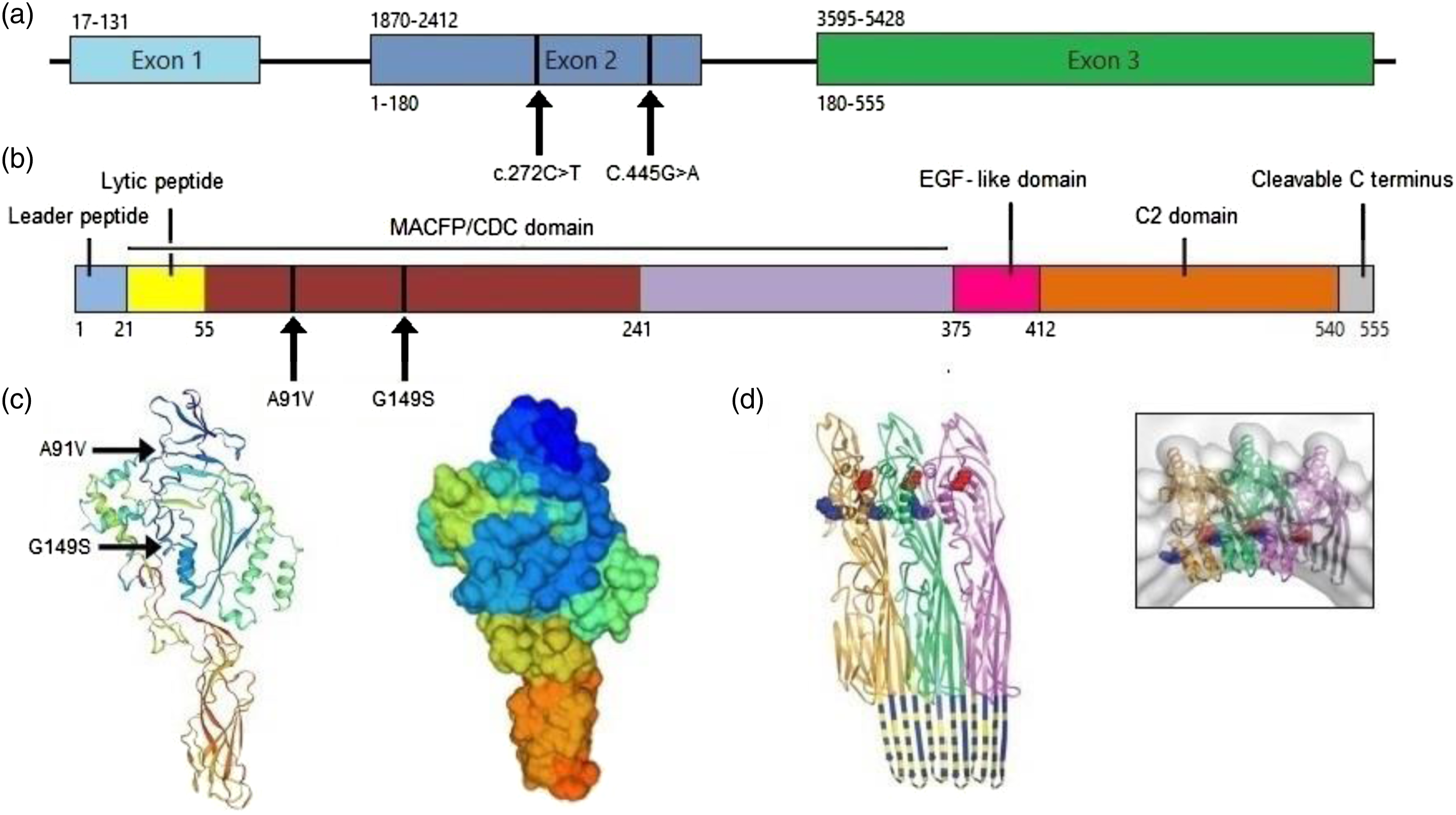
Figure 3, A molecular genetic study of the PRF1 gene was conducted in an asymptomatic biological son of the patient to analyse the presence/absence of the identified variants. The pathogenic variant c.445G>A, (p.Gly149Ser) was found in heterozygosis, supporting its trans position and co-segregation with the disease in the family. Unfortunately, it was not possible to genetically study the rest of the patient’s first-degree relatives. Proteomic and genomic organizations of perforin including the mutations found in the patient. (a) Diagram of PRF1 gene. Nucleotide position of each exon is indicated by number above the diagram, and amino acids encoded by the respective exons are indicated by number below the diagram. (b) Linear representation of matured perforin protein illustrating the different domains and amino acids of this molecule. (c) 3D rendering (cartoon, surface) of perforin structure (SWISS-MODEL Repository). (d) Model of perforin subunit packing in the pore seen from above and from inside. The amphipathic region is coloured by non-hydrophobic (blue) and hydrophobic (yellow) residues. From ‘The structural basis for membrane binding and pore formation by lymphocyte perforin’, by RHP Law et al., 2010, Nature, 468, p. 450.
8
Discussion
The finding of c.272C>T (p.Ala91Val) and c.445G>A (p.Gly149Ser) compound heterozygous variants in PRF1 could sustain the diagnosis of recessive FHL2 in our patient. Despite not having been able to carry out the molecular study in their parents and sisters, the presence of a single variant in their asymptomatic son supports its trans position and co-segregation with the disease in this family.
It has been estimated that FHL affects approximately 1 in 50,000 individuals worldwide, 14 but reported data is limited and varies between different studies and geographical regions: 0.12–0.15 cases/100,000 children/year (1.8/100,000 live births) in Sweden; 15 0.125 cases/100,000 individuals/year in Japan, 16 1 case per 100,000 children in Texas 17 and 7.5 cases/10,000 hospitalized children in Turkey, an increased rate due to the high frequency of consanguineous marriages in this region. 18 Of the five FLH subtypes, FLH2 is the most common, accounting for 20–40% of familial cases worldwide. 19 Its estimated frequency is between 12 and 750 cases per million children, although current incidence of each subtype varies in different populations and ethnic groups. 7 In general, FLH is diagnosed during the first year of life, having reported incidences of 1.1 cases/100,000 children <1 year, with a median age at diagnosis of 5.1 months. Even so, late-onset cases are increasingly studied and frequent, but there are few data about their incidence. 20
Numerous deletions, non-sense and missense germline mutations have been described in PRF1 gene associated with FHL2, mainly affecting the coding sequence. A high proportion of them are located in the MACPF/CDC domain, as in the concerning case. 21 Depending on their nature and location, different PRF1 mutations can lead to varying degrees of perforin expression, altering more or less severely the granule-dependent cytotoxicity pathway. Additionally, presentation of FHL2 correlates with other environmental and viral factors. For both reasons, not all patients with PRF1 variants will end up developing FLH2; and individuals with the same variants may present the disease with disparate clinical manifestations, variable involvement and different ages of presentation.22,23 In general, missense mutations affecting the C2 domain tend to be more deleterious and are associated with early onset FLH2. In contrast, MACPF/CDC domain mutations like the ones presented tend to be less damaging for the protein, excepting those that cause early truncation. 23 In such cases as in ours, patients with genotypes possibly predisposing to a greater probability of developing HLH may not develop symptoms for years, until infections (especially of viral origin), cancers or other external factors trigger the onset of the disease. This could explain why the FHL onset can be so delayed in some patients, as we highlight here.21,24,25
The c.272C>T (p.Ala91Val) variant has been previously associated with other perforin missense mutations in compound heterozygous patients with late onset disease, partial deficiency of perforin and residual but measurable cytotoxic activity in NK cells. 26 Late-onset FLH has a milder and more diverse clinical phenotype, which is easily misdiagnosed as other disorders including common variable immunodeficiency, non-Hodgkin lymphoma or an isolated neurologic disorder. 21 Ages of presentation vary between childhood and late adulthood, but to our knowledge, onsets >65 years have never been described in the literature. 27 These cases may be associated with milder and often recurrent HLH episodes with prolonged survival in absence of HSCT, unusual in the classical disease presentation. 21 In this line, our patient remained asymptomatic for 70 years, despite the drastic evolution suffered after the onset.
The minimum activity of perforin necessary to prevent FLH2 is unknown, but studies in mice suggest that 10–30% of cytotoxic T cells must express fully functional perforin to avoid symptoms associated with this disease. 28 Compared to our case, another report found in the literature with the same c.445G>A (p.Gly149Ser) variant but together with the c.116C>A (p.Pro39His) variant presented a much earlier age of onset (10 years), with absent NK function and lack of intracellular perforin in all cytotoxic cell types (complete deficiency). In contrast her asymptomatic mother, carrying the c.445G>A (p.Gly149Ser) variant in heterozygosis, showed a reduced proportion of certain perforin-expressing cells, with normal percentage of perforin-positive NKs and low perforin positivity in CD8+ and CD56+ T cells. 11 Also, it was described a case of an asymptomatic individual with the same two mutations as the patient of this paper, in whom they identify functional issues and suggest he may go on to develop symptoms in later life. 12 However, additional cases would still be required to cement this association.
The physiopathology of FLH2 is based on the existence of defects in the natural cytotoxicity of CTLs and NKs, although these cell counts remain normal. This leads to an uncontrolled expansion and activation of CTLs and macrophages, abnormal immune responses against pathogens (especially viruses) and excessive secretion of pro-inflammatory cytokines that result in the clinical and laboratory findings mentioned: IFNγ and TNFα trigger fever, cytokines suppress lipoprotein lipase leading to hypertriglyceridemia and macrophages produce elevated levels of ferritin that increase the concentration of plasminogen activator triggering hypofibrinogenemia. Some negative feedback mechanisms responsible for limiting the intensity and duration of the immune response against infections and aggressions are also affected in FLH2. These include destruction of antigen-presenting cells mediated by the perforin-granzyme and Fas-FasL systems of CTLs, cell death induced by the perforin-granzyme system and regulatory CD4-mediated elimination of expanded CD8. The consequent persistent exposure to antigens and permanence of hyperactivated CTLs generate a hyperinflammatory state that triggers the multiorgan dysfunction and FHL clinical manifestations.22,29
Conclusions
Familial hemophagocytic lymphohistiocytosis-2, caused by PRF1 mutations, is a rare and fatal autosomal recessive disorder in the absence of treatment that usually begins during the early childhood but can appear at any age, as shown in this case. Our results support the need of testing the presence of FLH-associated variants, not only for HLH patients in early childhood but also in adults with high suspicion of primary disease; since timely screening for mutated genes may be the key to early diagnosing, appropriate therapeutic decisions and correctly assess in other family members at risk. In this context, next-generation sequencing may be an efficient approach that could greatly help in early molecular diagnosis of FHL. In addition, bioinformatic tools help us to rapidly identify the functional consequences of disease-causing mutations, thus accelerating medical diagnosis of patients and providing timely and specific therapies that potentially improve life quality.
The clinical FHL presentation (prolonged fever, hepatosplenomegaly and cytopenias) and laboratory findings (hyperferritinemia, hypofibrinogenemia and hypertriglyceridemia) are the result of the hyperinflammatory state and cytotoxicity defects that arise from the absence/reduction of perforin levels. Current standard of treatment combines chemotherapy and immunotherapy (etoposide, cyclosporine, dexamethasone and intrathecal methotrexate) allowing the disease’s control in the majority of patients before performing HSCT, the only curative therapy available. Although much progress has been made in the knowledge of FLH2 in recent years, more studies are required to identify new variants, establish its real incidence in different ethnic groups and devise optimal diagnostic and therapeutic approaches for each patient.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Written informed consent was obtained from the patient.
Guarantor
NGR.
Contributorship
PSB, NGR, BMJ and SIA drafted and critically reviewed the manuscript with literature research. RAA, EGG and BMJ contributed to data collection and literature search. RGT helped to draft the manuscript. All authors read and approved the final manuscript.