
Abstract
Background
Deletions in the β-globin cluster are uncommon and cause thalassemia (thal) with hereditary persistence of fetal hemoglobin. They constitute a heterogenous group of disorders characterized by absent or reduced synthesis of adult hemoglobin (Hb A) and increased synthesis of fetal hemoglobin (Hb F). Although the clinical severity of these disorders are asymptomatic owing to the increased Hb F levels, the molecular basis is very heterogenous due to the large deletions in the β-globin cluster spanning both HBD and HBB genes. Here, we describe a Tunisian family carrying a novel deletion mutation causing (δβ)°-thalassemia.
Methods
The amounts of hemoglobin fractions were measured by capillary electrophoresis of hemoglobin. Amplification and sequencing of different regions on the β-gene cluster were performed by Sanger method.
Results
Family study and genetic analysis revealed a large deletion mutation in the β-globin cluster of 14.5 kb (NG_000,007.3:g. 58,253 to g.72837del14584) at the homozygous state in the patient and at heterozygous state at the other members of the family. This deletion removes the HBD and HBB genes.
Conclusions
In our knowledge, this new large deletion is described for the first time in the Tunisian population and in the world, designed Tunisian(δβ)0 in Ithanet database (IthaID: 3971). Therefore, it is important to identify the deletion leading to δβ-thalassemia carriers at the molecular level, to highlight the importance of recognizing the clinical features and implementing appropriate testing to clarify the diagnosis and manage the condition.
Keywords
Introduction
Thalassemia syndromes are the most common autosomal recessive genetic diseases and are responsible for significant morbidity and mortality worldwide. 1 β-thalassemia is often associated with point mutations or short deletions in the HBB gene, 2 but it rarely results from large gene deletion in β-globin cluster on chromosome 11. 3 Delta-beta thalassemia (δβ-thal) is caused by large heterogenous deletions in the β-globin cluster and characterized by the persistent expression of fetal hemoglobin during adult life.4,5 It is often associated with abolished or decreased expression of the adult HBD and HBB genes on the same chromosome 11; however, the HBG genes are rarely involved. 6 The hereditary persistent expression of fetal hemoglobin (HPFH) includes deletional HPFH and non-deletional HPFH (nd-HPFH), and the former is caused by large deletions in the region between the HBG and HBB genes. 7 However, it is not always possible to distinguish between HPFH and (δβ)--thalassemia because the distinction between them is subtle. Most cases of (δβ)-thalassemias described are heterozygotes, 8 only few cases with homozygous deletions in the β-globin cluster have been encountered thus far.9–11 The heterozygote cases have hematological parameters resembling those of a heterozygote for β°-thalassemia (β-thal) with severe microcytosis and hypochromia but with normal levels of Hb A2. 1 Here we describe a novel homozygous deletion in the β-globin cluster of 14.5 kb responsible for (δβ)°-thal in a Tunisian girl. This deletion is described for the first time in the Tunisian population.
Materials and methods
Patient
This study was approved by the National Ethics Committee of the Pasteur Institute of Tunis, Tunisia, and informed consent was obtained from all subjects participating in the study. A 7-year-old girl, offspring of consanguineous parents, was referred to our laboratory for investigation of chronic hemolytic mild anemia.
The history of the disease dates back to the age of 3 years of the patient. She consulted for the first time for anemia exploration without splenomegaly. Initial laboratory findings were as follows: Hb=8.5 g/dL, mean corpuscular volume (MCV)=73 fL, mean corpuscular Hb (MCH)=23 pg, serum iron=32 μmol/μL, and ferritin=151 ng/mL. Since then, a hematological follow-up was done at the age of 7 years and it showed a chronic anemia with Hb=8.7 g/dL, MCV=74.6 fL, and MCH=25.1 pg. Her physical examination revealed mucocutaneous pallor without stunted growth or facial dysmorphism. Discrete splenomegaly was observed with normal bone densitometry, Lactate dehydrogenase level and kidney function test. Laboratory data showed a reticulocyte count=85,000/mm3, hyperbilirubinemia (total bilirubin=45 μmol/l), and normal ferritinemia. The echo-abdominal examination revealed mild splenomegaly. Hemoglobin analysis showed 100% Hb F and an absence of Hb A and Hb A2. In view of these results, family investigation and molecular study were undertaken.
Hemoglobin analysis
Peripheral blood samples were collected from the family members for investigation. All subjects were initially screened by capillary electrophoresis (Capillarys Sebia 2 flex piercing) to determine the hemoglobin fractions.
Genetic analysis
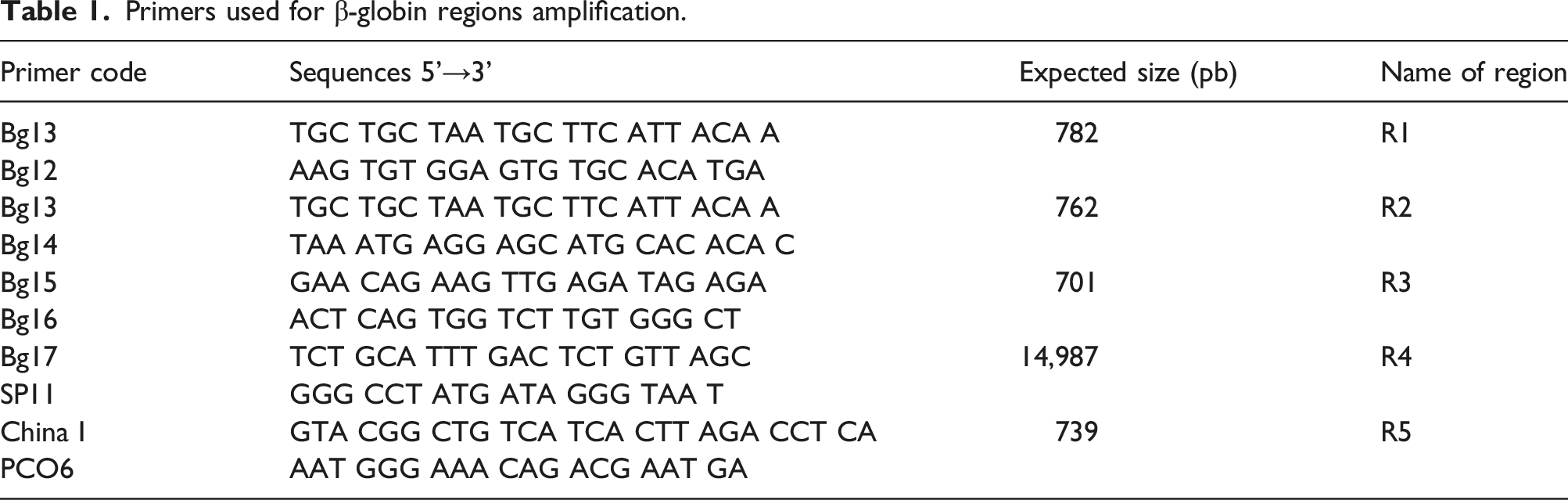
Genomic DNA was isolated from peripheral blood leukocytes by salting out extraction procedure. Simplex PCR was used in the first time to amplify a region (R5) of the HBB gene containing exon 1 and 2 according to the following protocol: initial denaturation at 94°C for 5 min, 30 cycles PCR (94°C for 1 min, 59°C for 1 min, 72°C for 1 min) and terminal elongation at 72°C. The unknown deletion was investigated by Gap-PCR. Specific regions corresponding to the human β-globin cluster (R1 to R4) was amplified (Figure 1(a)) using couples of primers listed in Table 1, as previously described.
12
PCR product of R4 region was sequenced in order to identify the breakpoint region. Summary of the molecular analysis of a novel β-gene cluster deletion (a) Locations of primers used in the β-globin gene cluster China I and PCO6 primers were used for the amplification of exon 1 and exon 2 of the HBB gene (R5). Bg12, Bg13, Bg14, Bg15, Bg16, Bg17, and SP11 primers were used for the precise localization of the deletion. (b) Sequencing result of the 403 pb fragment specific to the deletion in the patient. The amplification was realized with Bg17 and SP11 primers (R4). The direct sequencing shows the exact breakpoint of the deletion and suggests that the patient is homozygous for a 14.5 Kb deletion. Primers used for β-globin regions amplification.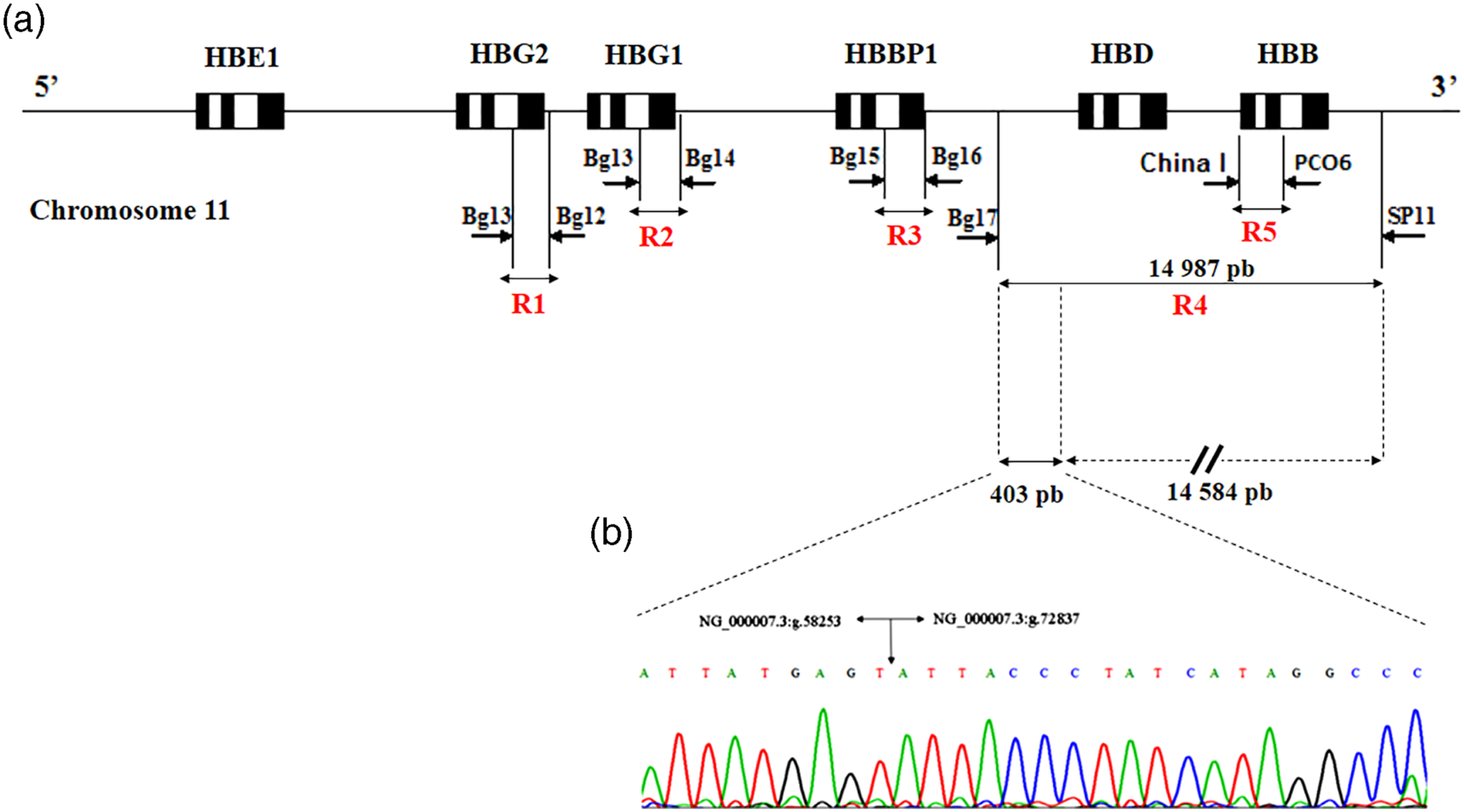
Gap Multiplex-PCR kit (QIAGEN® Multiplex PCR Kit, Germany) was used to detect the common deletion responsible for α-thalassemia (-α3.7, -α4.2, --SEA, --MED, --FIL, -α20.5, --THAI). 13 The PCR/Sequencing was performed to explore the HBA1 (α1-globin) and HBA2 (α2-globin) non-deletional mutations. 14
Results
Hematological data from the patient and her family are summarized in Figure 2. The patient has chronic hemolytic mild anemia and the red cells were hypochromic microcytic. Hemoglobin electrophoresis showed 100% Hb F in the proband, her parents and brother had the Hb F ranging from 11.6% to 15.4% and an Hb A from 82% to 85.9. HbA2 was within normal limits. Family pedigree and the hematologic and molecular data. The proband is indicated by an arrow.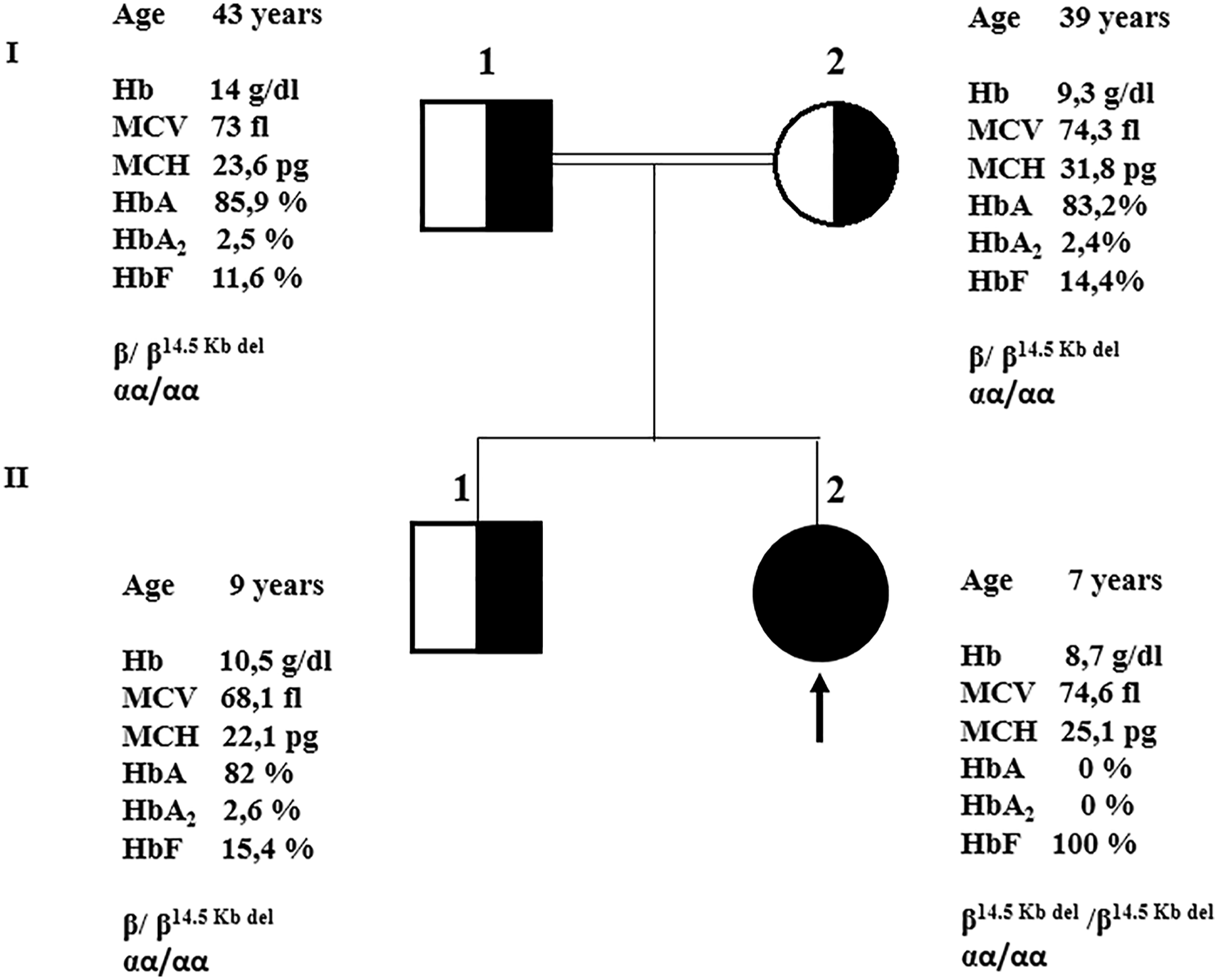
Molecular screening of HBA1 and HBA2 genes showed the absence of abnormalities. Molecular analysis of the β-gene cluster by Simplex PCR revealed an amplification of the R5 region among all family members except the proband, suggesting the presence of an unknown deletion of this region.
To determine the exact breakpoint location, we performed the GAP-PCR to amplify the specific regions on the β-globin cluster among the proband (Figure 1(a)). This screening strategy showed an amplification of the specific regions: R1, 2, and 3, except the R4 region containing HBD and HBB genes, which showed a smaller band of 403 pb, rather than the expected band of 14987 bp.
The sequencing analysis of the PCR product of R4 region (403 pb) showed the presence of a large deletion of 14584 pb covering the HBD and HBB genes, ranging from g.58253 to g.72837 (NG_000007.3:g. 58,253 to g.72837del14584) (Figure 1(b)). The proband was homozygous for this deletion. However, her brother and parents were heterozygous (Figure 2). This novel deletion has been submitted to the databases: Hbvar (https://globin.bx.psu.edu/cgi-bin/hbvar/counter) and Ithanet (https://www.ithanet.eu/).
Discussion
δβ-Thal results from large deletions removing the HBD and HBB genes among β-globin cluster on chromosome 11, with conservation of HBG gene.4,15,16 δβ-thal heterozygote have a modest elevation of Hb F (5–20%) and normal levels of Hb A2,5,17 while the homozygote forms are associated with microcytic hypochromic red cell indices and lead to thalassemia intermedia phenotype. 15
Hereditary persistence of fetal hemoglobin is caused by large deletions in the region between the HBG and HBB genes, 7 also by the occurrence of single point mutations in the promoter region of two γ-globin chains, which lead to an increased rate of γG and γA gene transcription. 18 Hereditary persistence of fetal hemoglobin heterozygotes are characterized by higher levels of Hb F (15–30%) with normal red cell indices and are asymptomatic at the homozygous state.5,17 The distinction between HPFH and δβ-thal is subtle and is made on clinical and hematologic grounds. 19
More than 50 types of δβ-thal or HPFH deletions have been reported, 20 which differ by position, size, and specific to ethnic origin of populations. 4 However, the homzygotes deletions among β-globin cluster are rarely reported.
Here, we report a new large homozygote deletion (14.5 kb) in the β-globin cluster that removes both the HBD and HBB genes, identified in a patient with 100% Hb F level. Exploration of the nucleotide sequences upstream and downstream of the junction breakpoints revealed a significant repeated sequence “ATTAC,” suggesting that the deletion was caused by a homologous recombination event.
This deletion leads to (δβ)°-thalassemia with hypochromic microcytic anemia (Hb=8.7 g/dl, MCV=74.6 fl, MCH=25.1 pg). This is the first report about the occurrence of a 14.5 kb (δβ)-globin genes homozygote deletion in Tunisian population and in the world, designed Tunisian(δβ)0 in Ithanet database (IthaID: 3971).
As the deletion has left the γ locus intact, Hb F continues to be produced at a high level to compensate the absence of normal adult hemoglobin. This elevation of Hb F explains that the clinical aspect was in favor of β-thalassemia intermedia since the patient has never been transfused and does not present a stunted growth.
In conclusion, this case illustrates the importance of the molecular diagnosis of thalassemia which can have clinical pictures of variable severity depending on the causal mutation. This disease analysis strategy presented here may be a valuable reference for similar problems permitting clinical geneticists to offer prenatal diagnosis to couples at risk.
Footnotes
Acknowledgements
This project is carried out under the MOBIDOC (mobilité pour les doctorants et post-doctorants) scheme, funded by the Ministry of Higher Education and Scientific Research through the PromESsE project (Projet de modernisation de l’Enseignement Supérieur en soutien à l’Employabilité) and managed by the ANPR (Agence Nationale de la Promotion de la Recherche scientifique).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
The study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Pasteur Institute’s Biomedical Ethics Committee, Tunisia (no.: 2018/11/I/LR16IPT07).
Guarantor
SM.
Contributorship
Study design: IM, IBF, FM and MO; Experimental work: MK, DC, and I.B; Interpretation of data: MK, IM, and HO; Manuscript preparation: MK, IM, and SM.