
Abstract
Background
Placental growth factor (PlGF) and soluble-fms-like tyrosine kinase 1 (sFlt-1) are biomarkers of placental function used to aid the diagnosis and prediction of pregnancy complications. This work verified the analytical performance of both biomarkers and provides preliminary diagnostic accuracy data to identify adverse pregnancy outcome in women with reduced fetal movement.
Methods
Verification of sFlt-1 and PlGF assays included a comparative accuracy assessment of 24 serum samples analysed at six different sites and laboratory-specific precision estimates. The sFlt-1/PlGF ratio was assessed in serum samples obtained prospectively from 295 women with reduced fetal movement ≥36 weeks’ gestation; diagnostic accuracy was evaluated using 2 × 2 tables and area under the receiver operator characteristic (AUROC) curve.
Results
Regression analysis showed that performance between sites was good with Passing-Bablok slopes ranging from 0.96 to 1.05 (sFlt-1) and 0.93 to 1.08 (PlGF). All sites had a mean bias <15%, although there was poorer agreement at the lowest PlGF concentrations. All within- and between-batch coefficients of variation were <10%. In 289 women with an appropriately grown fetus, an sFlt-1/PlGF ratio ≥38 had a sensitivity of 0.20 (95% confidence interval [CI] 0.07, 0.41), specificity of 0.88 (95% CI 0.83, 0.92) and AUROC curve of 0.58 (95% CI 0.47, 0.68) to identify adverse pregnancy outcome.
Conclusions
Analytical performance of the sFlt-1 and PlGF assays was comparable across different sites. The sensitivity of sFlt-1/PlGF to identify adverse pregnancy outcome in women with reduced fetal movement was considered acceptable, in the absence of other tests, to progress to a pilot randomized controlled trial.
Keywords
Introduction
Placental growth factor (PlGF) and soluble fms-like tyrosine kinase-1 (sFlt-1) are proteins present in maternal circulation that hold significant promise as biomarkers to aid the diagnosis and management of pregnancy complications, particularly those that relate to placental dysfunction.1–5 Research evaluating these biomarkers initially focused on detection and management of pre-eclampsia, 4 but more recent studies suggest they have potential value in identifying other complications including: prediction of small for gestational age (SGA) infants,5,6 fetal compromise after maternal presentation with reduced fetal movement (RFM) 7 and the need for intervention during labour, and compromise at the time of birth. 8 A systematic review of diagnostic test accuracy (DTA) studies found that PlGF was the most accurate biochemical test to identify pregnancies that would end in stillbirth with a diagnostic odds ratio of 49.2 (95% confidence interval [CI] 12.7, 191); however, this information comes from 5894 individuals, of whom only 16 had a stillbirth. 9 While this is an important avenue of exploration to prevent stillbirth, further studies are needed to increase understanding of test accuracy of PlGF in the prediction of adverse pregnancy outcome.
Measurement of sFlt-1 and PlGF utilizes immunoassays, the majority of which are automated. While there are a number of reports describing the diagnostic accuracy of these tests in a variety of clinical settings,3,5,10–12 descriptions of method performance are relatively limited. It is important to conduct such studies since the utility of these assays in clinical practice depends upon reproducible, accurate analysis. In addition, introduction of protocols using common thresholds requires an understanding of how analytical methods compare between laboratories. PlGF is bound in maternal blood by sFlt-1, and assays measure either unbound PlGF or the ratio of sFlt-1 to PlGF with the latter performing as well as, or better than, each biomarker alone in the diagnosis of pre-eclampsia.3,13,14 Results for the Elecsys® sFlt-1 and Elecsys® PlGF immunoassays (Roche Diagnostics, Germany) are comparable across a number of sites, 15 and similar analytical performance of these biomarkers has been demonstrated using other platforms including BRAHMS® Kryptor (ThermoFisher Scientific, Germany), 16 Alere Triage® PlGF test (Alere Inc., USA), DELFIA® Xpress PlGF 1–2-3 test (PerkinElmer; Finland) and ELISA (R&D Systems, USA).17,18 However, the methods have undergone further refinement and there is limited information regarding current performance including susceptibility to biotin interference which is a potential issue for some immunoassay systems.19–21
To understand method robustness in a wider clinical and laboratory context, we conducted a verification of analytical performance for the Elecsys® sFlt-1 and Elecsys® PlGF assays across sites participating in the ReMIT-2 trial. 22 This is a multicentre, randomized controlled, interventional pilot trial investigating whether including measurement of the sFlt-1/PlGF ratio following maternal presentation with RFM ≥36 weeks’ gestation with the aim of preventing adverse pregnancy outcome is feasible. This population was chosen for this study because RFM is associated with increased risk of adverse pregnancy outcomes and placental dysfunction,23–25 but management strategies based upon ultrasound scanning and induction of labour for RFM in all cases are associated with increased rates of obstetric intervention. 26 Our prior data indicating improved test performance with inclusion of PlGF assessment would potentially allow intervention to be focussed on women with RFM who were more likely to benefit from early induction. 7 Here, we report the performance of sFlt-1 and PlGF across six UK laboratory sites and assess potential confounding preanalytical factors including temperature stability and biotin interference. In addition, preliminary DTA data in this clinical context are presented, the objective of which was to evaluate the diagnostic accuracy of the sFlt-1/PlGF ratio in identifying a composite adverse pregnancy outcome in women presenting with RFM ≥36 weeks’ gestation.
Materials and methods
Preparation, distribution and analysis of samples
Blood samples were collected by venepuncture into clot activator gel tubes (Sarstedt; Nümbrecht, Germany), centrifuged at 3000 × g for 10 min, and the resultant serum split into aliquots and stored at –80°C. Frozen samples were sent on dry ice by same day courier and stored at –80°C at the receiving site until required. Sites were instructed to thaw samples immediately prior to analysis. All samples were analysed using the Elecsys® sFlt-1 and Elecsys® PlGF fully automated immunoassays on three different models of Roche Cobas® analyser (801, 602 and e411; Roche Diagnostics; Mannheim, Germany). Assays were run according to manufacturer’s instructions, and the sFlt-1/PlGF ratio was calculated for all samples.
Verification of sFlt-1 and PlGF analytical performance
Currently, there are no international reference materials to provide traceability of calibration of sFlt-1 or PlGF which contributes to a lack of gold standard analytical techniques to assess the accuracy of methods. Therefore, relative accuracy was addressed using anonymized residual serum blood samples routinely received in the Clinical Biochemistry Laboratory at the Royal Preston Hospital. For the study, 24 anonymized patient serum samples were distributed for analysis across six ReMIT-2 trial sites (Royal Preston Hospital, John Radcliffe Hospital, Sunderland Royal Hospital, St George’s Hospital, Royal Liverpool University Hospital and University Hospital of North Tees). The means of sFlt-1 and PlGF results for each sample were calculated and comparisons performed using Passing-Bablok regression analysis against the mean as a target. Additionally, the percent bias was determined for each site using the mean as the comparator. The clinical impact of any bias seen between sites was assessed by comparing the sFlt-1/PlGF ratios obtained from each sample against published thresholds for pre-eclampsia,3,10,27 with the mean value used as the comparator. These thresholds have been adapted for clinical use by the authors as follows: the risk of developing pre-eclampsia is considered low with a sFlt-1/PlGF ratio <38 (i.e. can be ruled-out for 1 week), intermediate with a ratio ≥38 to <85 (i.e. can be ruled-in within the next four weeks) and high with a ratio ≥85 (i.e. is a confirmed diagnosis).
Within-batch precision was determined on the 801 analyser at one site (Royal Preston Hospital) using three serum samples (n = 10). Between-batch precision was assessed using two levels of internal quality control (IQC) materials (PreciControl Multimarker, Roche Diagnostics, Mannheim, Germany) on the Cobas® analyser at all eight ReMIT-2 sites (Manchester Royal Infirmary and James Cook University Hospital in addition to those listed above). Between-batch precision data were collected to reflect real live usage over a time period to reach n = 20. The timeframe was dependent on the numbers done at each site and varied between three weeks and three months. For both assays, the lower limit of quantitation (LLOQ) was defined as the lowest measureable concentration with an acceptable precision and was assessed by analysing three low concentration samples in 10 separate runs. For both precision and LLOQ studies the mean, standard deviation (SD) and coefficient of variation (CV) were calculated.
The stability of sFlt-1 and PlGF was tested in five serum samples at room temperature over 24 h, at 4°C over 4 weeks, at –20°C over six months and over three freeze–thaw cycles. Biotin interference in the sFlt-1 and PlGF assays was determined at concentrations of 30, 60 and 500 ng/mL biotin. These were chosen to reflect serum biotin concentrations which might occur following ingestion of over-the-counter supplements or in patients taking mega doses for treating progressive multiple sclerosis.21,28 Two serum pools were spiked with increasing doses of biotin (Sigma-Aldrich; Gillingham, UK) according to methods described previously. 29 Working solutions were made up in distilled water, and the spiked amount was no more than 5% of the total serum volume. Additionally, distilled water was spiked as a control sample to account for dilutional or matrix effects. The mean percentage difference between the control sample and the spiked serum pools was then calculated.
DTA study
A preliminary DTA study was conducted to evaluate the sFlt-1/PlGF ratio (as the index test) for identifying adverse pregnancy outcome using maternal serum samples previously collected in three consecutive, separate studies run in a single tertiary UK maternity unit.7,30 Each of these had ethics approval for measurement of placental analytes (FEMINA Oldham Research Ethics Committee [REC] reference 08/H1011/83, recruitment took place from August 2009 – October 2019; FEMINA2 Greater Manchester West REC reference 11/NW/0650, recruitment took place from January 2012 to May 2014; FEMINA3 Greater Manchester East REC reference 16/NW/0481, recruitment started in August 2016 and is still ongoing), and written consent was obtained from all participants. Relevant eligibility criteria included women with a viable singleton pregnancy presenting with a primary complaint of maternal perception of RFM ≥36 weeks’ gestation and having had an ultrasound scan for fetal biometry, liquor volume and umbilical artery Doppler on presentation. Exclusion criteria included maternal age <18 years and fetus with a known congenital abnormality. Samples were analysed at site 6 using a Cobas® e411, and as this took place after birth, clinical care was not influenced by the sFlt-1/PlGF results, and assay results were not available when classifying pregnancy outcome.
Clinical and demographic details were collected from participants at presentation, along with ultrasound scan results for fetal biometry, liquor volume and umbilical artery Doppler. Delivery and pregnancy outcomes included gestational age at delivery, birthweight and gender. The target condition was a composite adverse pregnancy outcome defined as birthweight <5th centile (derived from individualized birth weight centiles 31 ), neonatal intensive care unit (NICU) admission, umbilical artery pH <7.1 at birth or stillbirth/neonatal death. Admission to NICU was regarded as any admission within the neonatal period regardless of indication. Where umbilical artery pH was missing, it was assumed to be normal since this would not be measured in normal healthy babies. The reference standards were each of the four individual components of the composite outcome. Since stillbirth alone as a measure of adverse pregnancy outcome would require a very large number of participants, a composite outcome was used similar to those employed in other perinatal trials.32,33 As sFlt-1 and PlGF were not measured until after birth, the clinicians determining whether the reference standard was present were not aware of the index test results. Similarly, those measuring the index test were unaware of whether the reference standard was present.
We aimed to include 410 participants in the DTA study to achieve a margin of error of 10% for sensitivity and 2.3% for specificity assuming that the prevalence of adverse pregnancy outcome would be 10%, sensitivity of the sFlt-1/PlGF ratio would be 80% and specificity would be 95%. The diagnostic accuracy of sFlt-1, PlGF and the sFlt-1/PlGF ratio was calculated using Stata Version 15 (StataCorp, College Station, USA). Performance of the sFlt-1/PlGF ratio was assessed using predefined levels of 38 (determined to be the optimal level to diagnose pre-eclampsia 3 ) and 112 (95th centile value >37 weeks’ gestation). The diagnostic accuracy of currently used tests including estimated fetal weight (EFW) <10th centile, and oligohydramnios (defined by gestation-specific centile for amniotic fluid index or maximal pool depth) as well as EFW <10th centile or sFlt-1/PlGF ratio ≥38 were also examined. Sensitivity, specificity, and positive and negative predictive values were calculated along with the area under the receiver operator characteristic (AUROC) curve for the sFlt-1/PlGF ratio; 95% CIs were calculated for test performance characteristics. Any instances of indeterminate or missing sFlt-1/PlGF ratios, or where all components for the adverse pregnancy outcome were indeterminate or missing, were excluded from the analysis.
Results
Verification of sFlt-1 and PlGF analytical performance
Results for sFlt-1, PlGF and the sFlt-1/PlGF ratio from 24 samples analysed at each site were compared using Passing-Bablok analysis with the mean of all sites as the comparator. For all sites and assays, this gave slopes ranging from 0.89 to 1.08 and intercepts ranging from –6.52 to 9.13 pg/mL (Table 1). In addition, percent bias for sFlt-1, PlGF and the sFlt-1/PlGF ratio from each site was plotted against the mean of all results which showed that for both biomarkers, most sites had a mean bias of <10% (Figure 1). The only exception was laboratory 6 which had a mean bias for PlGF of –10.2% (Figure 1(b)), primarily due to greater variability at concentrations below 25 pg/mL when bias values ranged from –56 to 24% (Figure 1(b)). The variability at low PlGF concentrations impacted on the bias of the calculated sFlt-1/PlGF ratio; however, over 70% of the sFlt-1/PlGF ratio results had a bias <10% (Figure 1(c)). Assessment of the clinical impact of this bias performed on individual sFlt-1/PlGF ratio results (n = 144) indicated that two samples (1.4%) would have been classified as having an intermediate risk of developing pre-eclampsia where the consensus mean was low risk.
Passing-Bablok slope and intercept results for sFlt-1, PlGF and sFlt-1/PlGF ratio at each site.
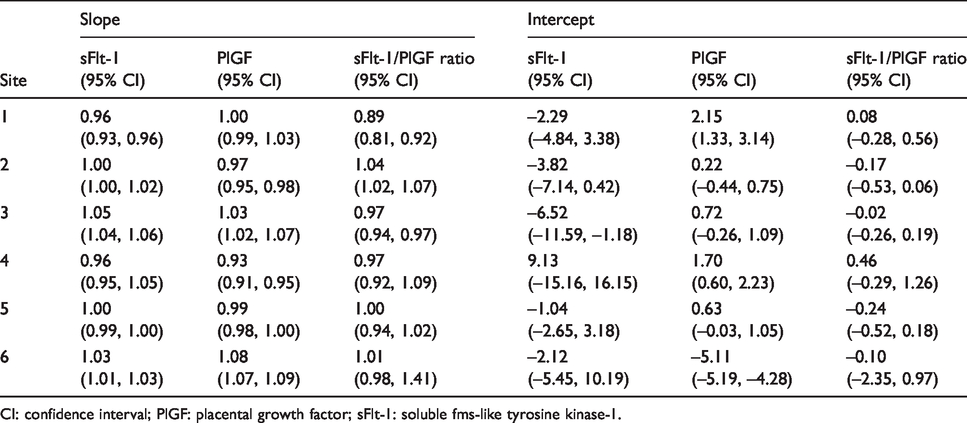
CI: confidence interval; PlGF: placental growth factor; sFlt-1: soluble fms-like tyrosine kinase-1.

Percent bias for each site for sFlt-1, PlGF and sFlt-1/PlGF ratio (n = 24). The individual site percent bias is plotted against the mean of all sites for: (a) sFlt-1; (b) PlGF; (c) sFlt-1/PlGF ratio. Site 1 (801 Cobas® analyser);  Site 2 (602 Cobas® analyser);
Site 2 (602 Cobas® analyser);  Site 3 (e411 Cobas® analyser);
Site 3 (e411 Cobas® analyser);  Site 4 (801 Cobas® analyser);
Site 4 (801 Cobas® analyser);  Site 5 (602 Cobas® analyser);
Site 5 (602 Cobas® analyser);  Site 6 (e411 Cobas® analyser).
Site 6 (e411 Cobas® analyser).
Within-batch precision CVs for sFlt-1 were 1.13%, 0.53% and 0.63% at 82.1 pg/mL, 1020.6 pg/mL and 13393.1 pg/mL, respectively (n = 10). Within-batch precision CVs for PlGF were 1.20%, 0.69% and 1.04% at 14.5 pg/mL, 143.9 pg/mL and 1319.3 pg/mL, respectively (n = 10). Between-batch precision estimates were generally <5% at both levels of IQC material, mean CVs for all eight sites being 4.6% (Low QC) and 4.9% (High QC) for sFlt-1 along with 4.2% (Low QC) and 4.3% (High QC) for PlGF (Table 2). Precision estimates at one site with the e411 analyser were much higher (>20% for sFlt-1 and around 10% for PlGF), although this was not instrument type related, as precision at other sites with the e411 analyser was <5% for both biomarkers at the concentrations assessed.
Between-batch method precision.
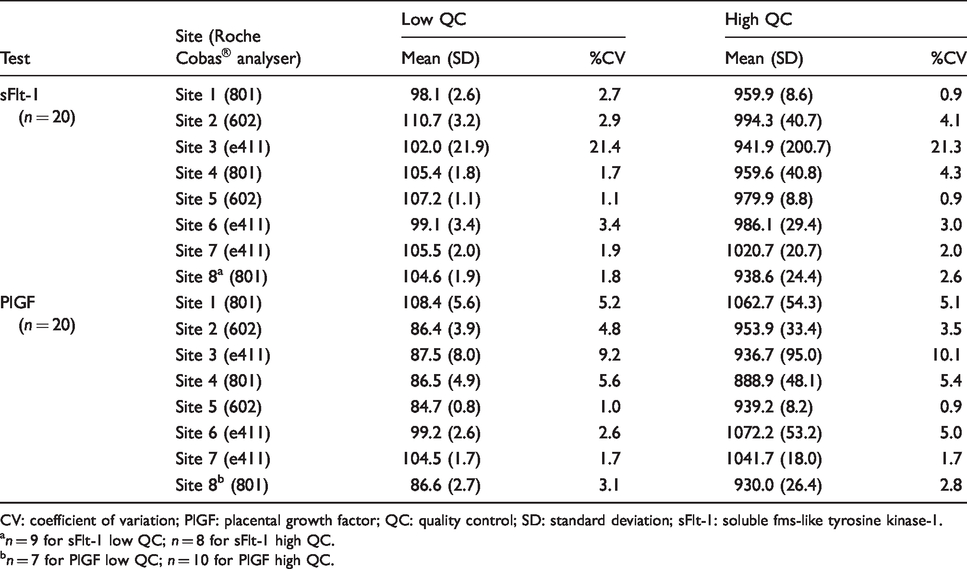
CV: coefficient of variation; PlGF: placental growth factor; QC: quality control; SD: standard deviation; sFlt-1: soluble fms-like tyrosine kinase-1.
an = 9 for sFlt-1 low QC; n = 8 for sFlt-1 high QC.
bn = 7 for PlGF low QC; n = 10 for PlGF high QC.
For the PlGF LLOQ, mean concentrations analysed were 5.2 pg/mL, 8.9 pg/mL and 10.7 pg/mL which gave CVs of 9.05%, 4.31% and 6.24%, respectively. The LLOQ studies for sFlt-1 were performed at mean concentrations of 12.1 pg/mL, 13.3 pg/mL and 21.5 pg/mL which resulted in CVs of 7.60%, 4.24% and 4.92%, respectively.
sFlt-1 and PlGF were demonstrated to be stable in serum when stored at –20°C for six months (Figure 2(a)), when stored at 4°C for 14 days (Figure 2(b)) and when stored at room temperature for 24 h (Figure 2(c)). All serum samples tested had a mean difference from baseline of <10%. Additionally, both sFlt-1 and PlGF were stable over three freeze–thaw cycles (Figure 2(d)).
Stability of sFlt-1 and PlGF in serum over various timeframes (n = 5). Stability of sFlt-1 and PlGF: (a) at –20°C over six months; (b) at 4°C over 14 days; (c) at room temperature over 24 h; (d) over three freeze–thaw cycles. sFlt-1; PlGF; error bars are 95% confidence intervals.
The addition of biotin to pooled serum samples resulted in a reduction of >10% from the spiked control sample in the PlGF assay at biotin concentrations of 30, 60 and 500 ng/mL (Supplement Figure 1). The effect was related to the concentration of biotin, with 500 ng/mL resulting in a greater than –80% difference. With sFlt-1, interference was not as significant with biotin concentrations of 30 and 60 ng/mL having <10% bias (Supplement Figure 1). However, at 500 ng/mL, the difference was –79% from the baseline sample.
DTA study
Across the three studies, samples from 423 participants were analysed (Supplement Figure 2). Three hundred and eighteen participants met the relevant inclusion criteria for the DTA study (≥36 weeks’ gestation). Twenty-three were excluded due to the EFW being <10th centile at presentation (i.e. the fetus already had a diagnosis of being SGA), since this would be an indication for delivery after 37 weeks’ gestation and therefore not meet the inclusion criteria for ReMIT-2, 22 and a further six had insufficient serum to conduct assays for both biomarkers which gave 289 participants with both sFlt-1 and PlGF results to calculate the ratio of both biomarkers. Maternal characteristics at baseline for all 318 participants are shown in Supplement Table 1; the mean age of participants was 29.2 years (standard deviation [SD] 5.5), mean body mass index (BMI) was 26.4 (SD 5.4) and just over half (54%) were in their first pregnancy.
In 295 participants presenting with RFM ≥36 weeks’ gestation whose babies were ≥10th EFW centile, 26 (8.8%) had the target condition (composite adverse pregnancy outcome); 2.4% had a birthweight <5th centile, 4.1% were admitted to NICU, 3.7% had an umbilical artery pH <7.1 at birth, and there were no stillbirths/neonatal deaths. Three babies had both low pH and NICU admission and one further baby had birthweight <5th centile and NICU admission. In babies who were ≥10th EFW centile and had results for both biomarkers (n = 289), the sFlt-1/PlGF ratio at a threshold of ≥38 had a sensitivity of 0.20 (95% CI 0.07, 0.41) and a specificity of 0.88 (95% CI 0.83, 0.92) to predict the composite adverse pregnancy outcome (Table 3). Applying a threshold of ≥112 for the sFlt-1/PlGF ratio had a sensitivity of 0.04 (95% CI 0.00, 0.20) with a specificity of 0.99 (95% CI 0.97, 1.00) (Table 3). The AUROC curve of the sFlt-1/PlGF ratio to identify adverse pregnancy outcomes in these women was 0.58 (95% CI 0.47, 0.68; Figure 3). For comparison, ultrasound EFW <10th centile alone (n = 318) had a sensitivity of 0.24 (95% CI 0.11, 0.41) and a specificity of 0.95 (95% CI 0.91, 0.97), isolated oligohydramnios (n = 295) had a sensitivity of 0.12 (95% CI 0.02, 0.30) and a specificity of 0.85 (95% CI 0.80, 0.89) and either EFW <10th centile or sFlt-1/PlGF ≥38 (n = 312) had a sensitivity of 0.39 (95% CI 0.23, 0.58) and a specificity of 0.83 (95% CI 0.78, 0.87; Table 3) to predict the composite adverse pregnancy outcome. No significant adverse events occurred as a result of taking the blood sample to perform the sFlt-1/PlGF test, or from measuring the birthweight or umbilical artery pH.
Diagnostic accuracy of ultrasonographic and biomarker tests to identify composite adverse pregnancy outcome.
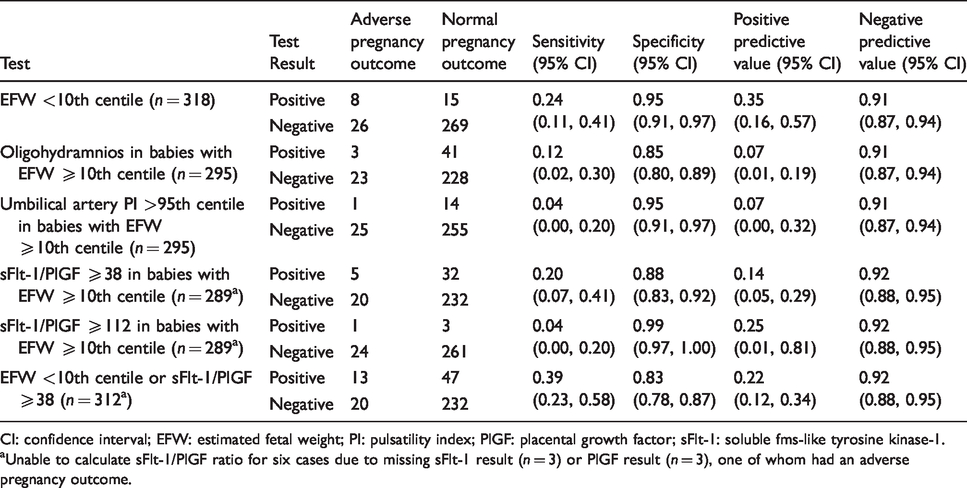
CI: confidence interval; EFW: estimated fetal weight; PI: pulsatility index; PlGF: placental growth factor; sFlt-1: soluble fms-like tyrosine kinase-1.
aUnable to calculate sFlt-1/PlGF ratio for six cases due to missing sFlt-1 result (n = 3) or PlGF result (n = 3), one of whom had an adverse pregnancy outcome.
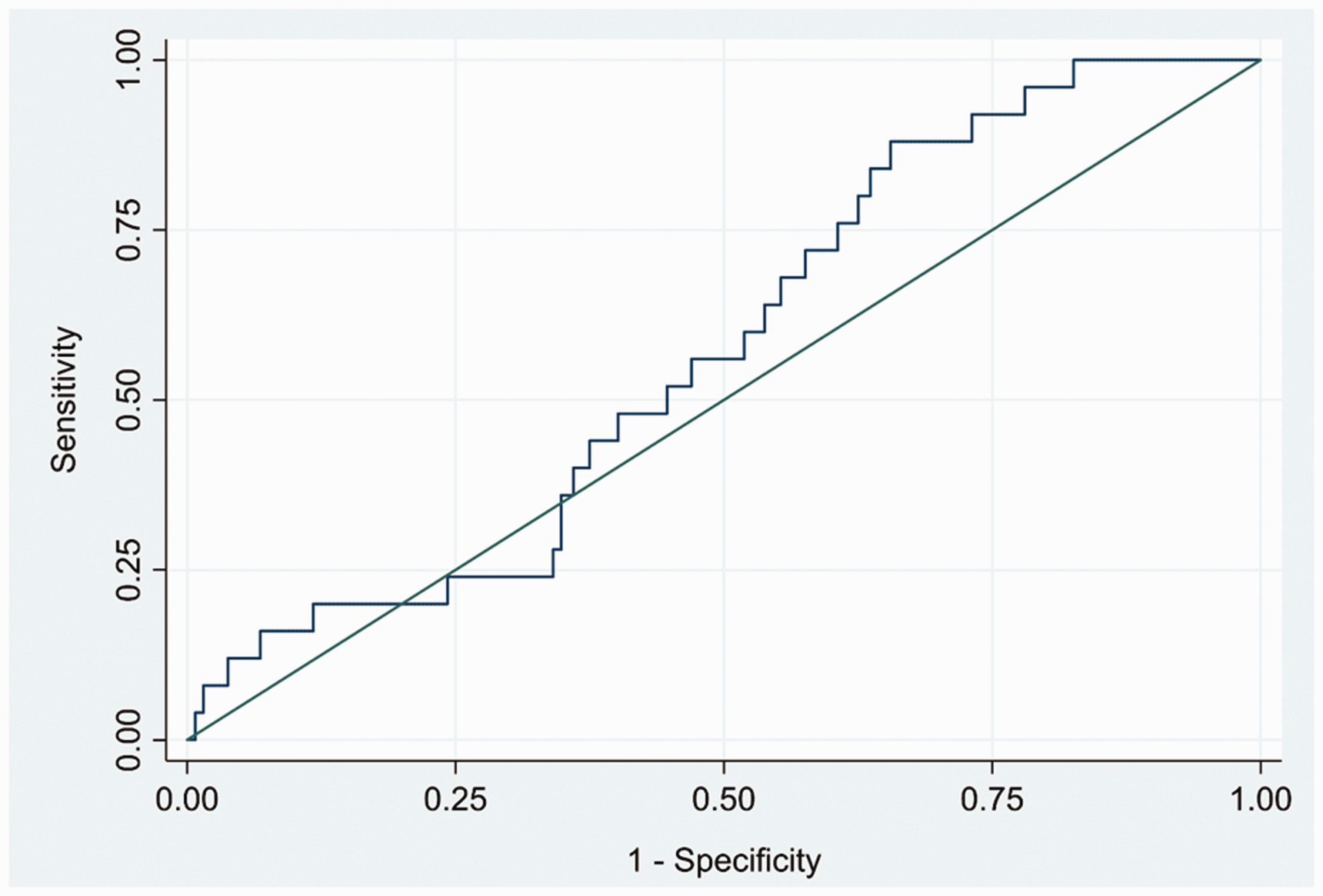
Receiver operator characteristic curve for sFlt-1/PlGF ratio to predict composite adverse pregnancy outcome. AUROC 0.58 (95% CI 0.47, 0.68).
Discussion
The potential for biomarkers of placental function to help detect pregnancy complications and aid diagnosis is an area of clinical practice that is regaining momentum after a period of initial study in the 1970s and 1980s. The majority of recent work has focused on the role of PlGF or the sFlt-1/PlGF ratio in the diagnosis of pre-eclampsia which has led to the National Institute for Health Care and Excellence (NICE) issuing guidance on the use of these biomarkers in suspected pre-eclampsia. 34 As pre-eclampsia has its origins in placental dysfunction, there is also interest in utilizing placentally-derived biomarkers such as PlGF for other conditions related to poor placental function, e.g. fetal growth restriction or pregnancies that are likely to end in stillbirth 6 or fetal and neonatal compromise resulting from labour. 8
Although there are various methods available to quantify sFlt-1 and PlGF, NICE guidance for pre-eclampsia specifically supports the use of the Elecsys® sFlt-1 and Elecsys® PlGF assays (Roche Diagnostics; Germany). 34 Therefore, we chose to use the Elecsys® system in this work and assessed test reproducibility of both assays across six different sites. There are currently no defined or published acceptance criteria for these biomarkers based on biological variability, but we consider the performance reported here acceptable across most of the concentration range tested. Performance of the PlGF assay at concentrations <25 pg/mL was more variable, and caution is advisable when interpreting results at this lower limit. This may be due to these values being at the very lowest PlGF concentrations of a very wide analytical range (3–10,000 pg/mL, limit of quantification 10 pg/mL) with a potentially greater impact of calibration curve fitting models. However, the absolute differences were small and are all below PlGF concentrations that should raise concerns in clinical practice; for example, NICE suggest a threshold of 100 pg/mL to define abnormality 34 in the context of pre-eclampsia. If a ratio approach is used, there is potential to increase its variability in the instances where PlGF concentrations are below 25 pg/mL, and this also needs to be considered in interpretation. However, when the individual sFlt-1/PlGF ratios obtained by the six sites in this study were compared with pre-eclampsia diagnosis thresholds, <2% would have changed classification. This suggested that the variable performance at low concentrations of PlGF described here would not have a major effect on clinical outcomes.
Between-batch precision was acceptable for both assays with the mean CV assessed at eight sites being <5%. However, one outlier was noted at site 3 which had significantly higher imprecision, in particular for sFlt-1, than the other sites. Most of the sites were in the method verification stage of using these biomarkers, and therefore the observed precision at site 3 may have been compromised by either inconsistent handling of IQC materials or inexperience in using the assays. The most experienced laboratory (site 6) had noted that IQC performance decreased following initial opening of the bottle, even when it was kept refrigerated, and their standard practice changed to freezing aliquots of IQC material which improved observed precision. Further evaluation of sFlt-1 and PlGF stability in IQC materials is needed, particularly as the instability noted is inconsistent with the performance of these biomarkers in serum samples. This observation also highlights the need for the development of independent third party IQC materials. Additionally, the clinical impact of bias and imprecision for these markers needs further consideration, and this should be explored as external quality assessment schemes develop. Precision studies show that both sFlt-1 and PlGF assays can be acceptably quantitated down to the lowest concentrations tested with %CVs less than 10. We report acceptable precision and therefore a LLOQ at 12.1 pg/mL for sFlt-1 and 5.2 pg/mL for PlGF which highlights an improvement in the levels stated in the manufacturer’s product sheets (15 pg/mL and 10 pg/mL, respectively).
Both biomarkers also showed stability across a range of storage conditions and timeframes. This was an important consideration for samples collected in the ReMIT-2 trial since they are stored at –20°C or lower for a maximum of six months prior to central analysis of sFlt-1 and PlGF which is being conducted as a confirmatory measure of reliability. 22 Biotin interference with the sFlt-1 assay was limited at lower concentrations, but a greater effect was seen with the PlGF assay at 60 ng/mL. Both assays showed a much larger difference with biotin at a higher concentration of 500 ng/mL. Pregnancy multivitamin tablets taken once daily contain around 50–150 µg of biotin, so it is unlikely that pregnant women using such products will have blood concentrations high enough to interfere with the sFlt-1 and PlGF assays. However, biotin is available as a single vitamin supplement at doses up to 10 mg once per day and this much higher concentration could potentially cause issues with accurate measurement of both biomarkers.
These analytical performance findings agree with previously published manufacturer’s data 15 and have provided reassurance that the sFlt-1 and PlGF results generated in the ReMIT-2 trial and other multicentre clinical evaluations will be robust. This is particularly important since there is currently no External Quality Assessment scheme available in the UK for sFlt-1. It is therefore advisable to conduct similar analytical performance and stability research alongside any future trials using these biomarkers. In addition, the potential issue of biotin interference should be given due consideration in any research using the Elecsys® sFlt-1 and Elecsys® PlGF assays.
For the DTA study, although the sensitivity of the sFlt-1/PlGF ratio ≥38 to detect an adverse pregnancy outcome in women with RFM was modest and lower than that used to diagnose pre-eclampsia, it was comparable to, or better than, other methods currently used in clinical practice following maternal presentation with RFM, e.g. ultrasound fetal biometry, liquor volume and umbilical artery Doppler >95th centile (Table 3). The combination of EFW <10th centile or sFlt-1/PlGF ratio ≥38 was included for comparison and did improve sensitivity; however, as EFW <10th centile would be an indication for immediate delivery, this would not meet the inclusion criteria for the ReMIT-2 trial. 22 Using a threshold of ≥112 for the sFlt-1/PlGF ratio did not improve prognostic performance. In common with analysis of the whole FEMINA2 data-set, addition of PlGF measurement improved the sensitivity to detect adverse pregnancy outcome above use of ultrasound alone. 7 Importantly, views on this level of sensitivity and specificity were sought from a patient and public involvement group (personal communication) and the independent Trial Steering Committee. These groups agreed that the increased sensitivity of adding sFlt-1/PlGF testing to currently available regimens without a significant reduction in specificity would aid the clinical management of women at risk of an adverse pregnancy outcome, and was therefore deemed an appropriate test to investigate further in a pilot trial. This opinion was strengthened by the fact that, in the absence of the sFlt-1/PlGF test, women with RFM would be randomized to continue with normal care in their pregnancy. 22
The AUROC curve of 0.58 observed in this study indicates that the sFlt-1/PlGF ratio is slightly better than chance to discriminate between women with and without composite adverse pregnancy outcomes. Overall, our findings for the sFlt-1/PlGF ratio are in agreement with other analyses which suggests that PlGF measurement modestly improves the detection of adverse pregnancy outcome although this may differ for specific adverse outcomes. Analysis of a cohort of 3953 participants found that a combination of EFW, PlGF and uterine artery Doppler pulsatility index had a sensitivity of 0.63 for SGA births. 35 In contrast, a combination of EFW and sFlt-1 had a sensitivity of 0.15 for emergency Caesarean section for fetal compromise during labour, and adverse outcomes after birth (low pH, Apgar score ≤7 at 5 min, NICU admission) were not predicted by any single or combination of measurements or biochemical or biophysical predictors. 35 Our recent systematic review of DTA studies found that PlGF had the best predictive accuracy for identifying a pregnancy that ends in stillbirth, but that its performance was not as good as EFW alone in prediction of a SGA infant. 9 Therefore, further studies are needed to determine which biomarkers most accurately identify fetal and neonatal compromise and whether combinations of biomarkers are more successful than when used in isolation.
The current study limitations relate to sample size for both laboratory comparisons and diagnostic accuracy and the unblinded nature of the ultrasound scan findings. For the latter issue, this means that clinicians are aware of the results for EFW or abnormal liquor volume and umbilical artery Doppler which may lead to intervention, thereby avoiding a potential adverse pregnancy outcome and confounding the observed diagnostic performance of these tests. Although restricted by sample size, the laboratory study represents real-world data from routine diagnostic laboratories and is the first data we are aware of showing how these methods perform across multiple sites in this setting. The planned sample size for the DTA study was not met, meaning that the confidence intervals for the sensitivity and specificity of the sFlt-1/PlGF ratio to identify adverse pregnancy outcomes were slightly wider than planned. In addition, although modest improvements using the sF1t-1/PlGF ratio were observed in this study, the confidence intervals for the sensitivity and specificity overlapped with the other tests, meaning a larger sample size would be required to identify whether the sF1t-1/PlGF ratio is better than the other tests. To address this, the FEMINA3 trial is ongoing, and additional samples for DTA studies will also be obtained from the control arm of the ReMIT-2 trial. 22 Importantly, future clinical studies also need to address the impact of ethnicity, BMI and presence of co-morbidities on the test performance of the sFlt-1/PlGF ratio.
The DTA study is strengthened, as samples were obtained from a single tertiary UK maternity unit serving a population with a diverse demographic and ethnic background, plus eligibility criteria were kept simple to give a degree of generalizability. In addition, the investigators determining the index test and those determining the reference standard were blinded to each other’s results. Furthermore, this DTA analysis reports similar findings to a study using PlGF alone (rather than the sFlt-1/PlGF ratio) in low-risk pregnancies predicting fetal or neonatal compromise, with AUROCs between 0.51 and 0.67 for different definitions of fetal and neonatal compromise. 36
To the best of our knowledge, this is the first report of sFlt-1/PlGF DTA results in women with RFM in late pregnancy and is a pragmatic initial exploration of test characteristics for these assays in this population. Knowledge of test characteristics and technical variation is particularly important when applying a threshold to initiate management or treatment as small changes in absolute values can modify the ratio. The sFlt-1/PlGF threshold of ≥38 is being assessed further in the ongoing ReMIT-2 pilot trial to identify adverse pregnancy outcome in women with RFM, the results of which will help determine whether a much larger trial using these biomarkers in such a context is warranted.
Supplemental Material
ACB911993 Supplemental Material - Supplemental material for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement
Supplemental material, ACB911993 Supplemental Material for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement by Lindsay K Armstrong-Buisseret, Shonagh Haslam, Tim James, Lucy Bradshaw and Alexander EP Heazell in Annals of Clinical Biochemistry
Supplemental Material
ACB911993 Supplemental Material2 - Supplemental material for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement
Supplemental material, ACB911993 Supplemental Material2 for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement by Lindsay K Armstrong-Buisseret, Shonagh Haslam, Tim James, Lucy Bradshaw and Alexander EP Heazell in Annals of Clinical Biochemistry
Supplemental Material
ACB911993 Supplemental Material3 - Supplemental material for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement
Supplemental material, ACB911993 Supplemental Material1 for Verification of placental growth factor and soluble-fms-like tyrosine kinase 1 assay performance in late pregnancy and their diagnostic test accuracy in women with reduced fetal movement by Lindsay K Armstrong-Buisseret, Shonagh Haslam, Tim James, Lucy Bradshaw and Alexander EP Heazell in Annals of Clinical Biochemistry
Footnotes
Acknowledgements
The trial Sponsor is the University of Manchester and the trial co-ordinating centre is Nottingham Clinical Trials Unit. The measurement of the sFlt-1/PlGF ratio within this study was supported by Roche Diagnostics International Ltd; however, Roche had no involvement in the design and conduct of the research or interpretation of the results. The authors would like to thank the following individuals for analysing samples at their sites and providing data: Karen Perkins (Royal Preston Hospital), Caroline Addison (Sunderland Royal Hospital), Liz Okokon (St George’s Hospital, London), Suzannah Phillips (Royal Liverpool University Hospital), Ian Smith (John Radcliffe Hospital, Oxford), Helen Verrill (University Hospital of North Tees), Katharine Hayden and Michelle Nuttall (Manchester Royal Infirmary) and Steven Liggett and Andrew Teggert (James Cook University Hospital, Middlesbrough).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project is funded by the National Institute for Health Research (NIHR) Clinical Scientist Fellowship Award (CS-2013–13-009). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Ethical approval
Guarantor
AEPH.
Contributorship
AEPH conceived the study, gained funding and ethical approval and was involved in patient recruitment. AEPH and LKAB were involved in protocol development. SH and TJ were involved in method development. LB was involved in statistical analysis. LAB wrote the first draft of the article. All authors reviewed and edited the article and approved the final version of the article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.