
Abstract
Proprotein convertase subtilisin kexin 9 (PCSK9) is a serine protease with a key role in regulating plasma low-density lipoprotein (LDL) concentration. Since its discovery via parallel molecular biology and clinical genetics studies in 2003, work to characterize PCSK9 has shed new light on the life-cycle of the low-density lipoprotein receptor and the molecular basis of familial hypercholesterolaemia. These discoveries have also led to the advent of the PCSK9 inhibitors, a new generation of low-density lipoprotein cholesterol (LDL-C) lowering drugs. Clinical trials have shown these agents to be both safe and capable of unprecedented reductions in LDL-C, and it is hoped they may herald a new era of cardiovascular disease prevention. As such, the still evolving PCSK9 story serves as a particularly successful example of translational medicine. This review provides a summary of the principal PCSK9 research findings, which underpin our current understanding of its function and clinical relevance.
Introduction
Familial hypercholesterolaemia (FH) is one of the most common inherited metabolic disorders with an estimated prevalence of between 1:500 and 1:200.1 If untreated, FH is characterized by high concentrations of plasma low-density lipoprotein cholesterol (LDL-C), early onset coronary heart disease (CHD) and hypercholesterolaemic stigmata such as tendon xanthoma or corneal arcus. Fortunately, the early-onset CHD associated with heterozygous FH (heFH) can be prevented if identified and treated early with LDL-C lowering drugs; in fact there is strong evidence that statin therapy reduces the risk of CHD in heFH to a level comparable with the general population. 2
FH was probably first described as an inherited clinical syndrome in 1938 by the Norwegian physician, Carl Müller. 3 His studies of around 17 families in Oslo describe many of its hallmark features including early-onset CHD and tendon xanthoma and the dominant nature of its inheritance. However it was not until 1964 when Kachadurian formally demonstrated the existence of both heterozygous and homozygous forms of FH 4 through genetic studies in Lebanese FH families, that its autosomal co-dominant nature was definitively determined. FH is now understood to be caused by mutations in one of three separate genes (Note: A very rare autosomal recessive form of hypercholesterolaemia also exists. This is caused by loss-of-function mutations in the ARH gene which encodes the low-density lipoprotein receptor adaptor protein (LDLRAP), a protein required for normal LDL-receptor mediated endocytosis. Deficient LDLRAP thereby leads to impaired clearance of LDL from the circulation.): low-density lipoprotein receptor (LDLR), apolipoprotein B (ApoB), the ligand for LDLR, and proprotein convertase subtilisin kexin 9 (PCSK9). Their corresponding proteins each perform crucial roles in the clearance of plasma LDL-C by hepatocytes (Figure 1). Ground-breaking work in the 1970s by Brown and Goldstein which investigated the biological basis for FH,5,6 resulted in the discovery of the LDL receptor and, together with an extensive body of related later work, led to Brown and Goldstein being awarded the 1985 Nobel Prize for Physiology or Medicine. In the 1980s, Grundy and co-workers showed that the FH phenotype could also arise from mutations in ApoB.7–9 However, it was not until the beginning of the new millennium some 20 years later that the third FH gene, PCSK9, was identified.
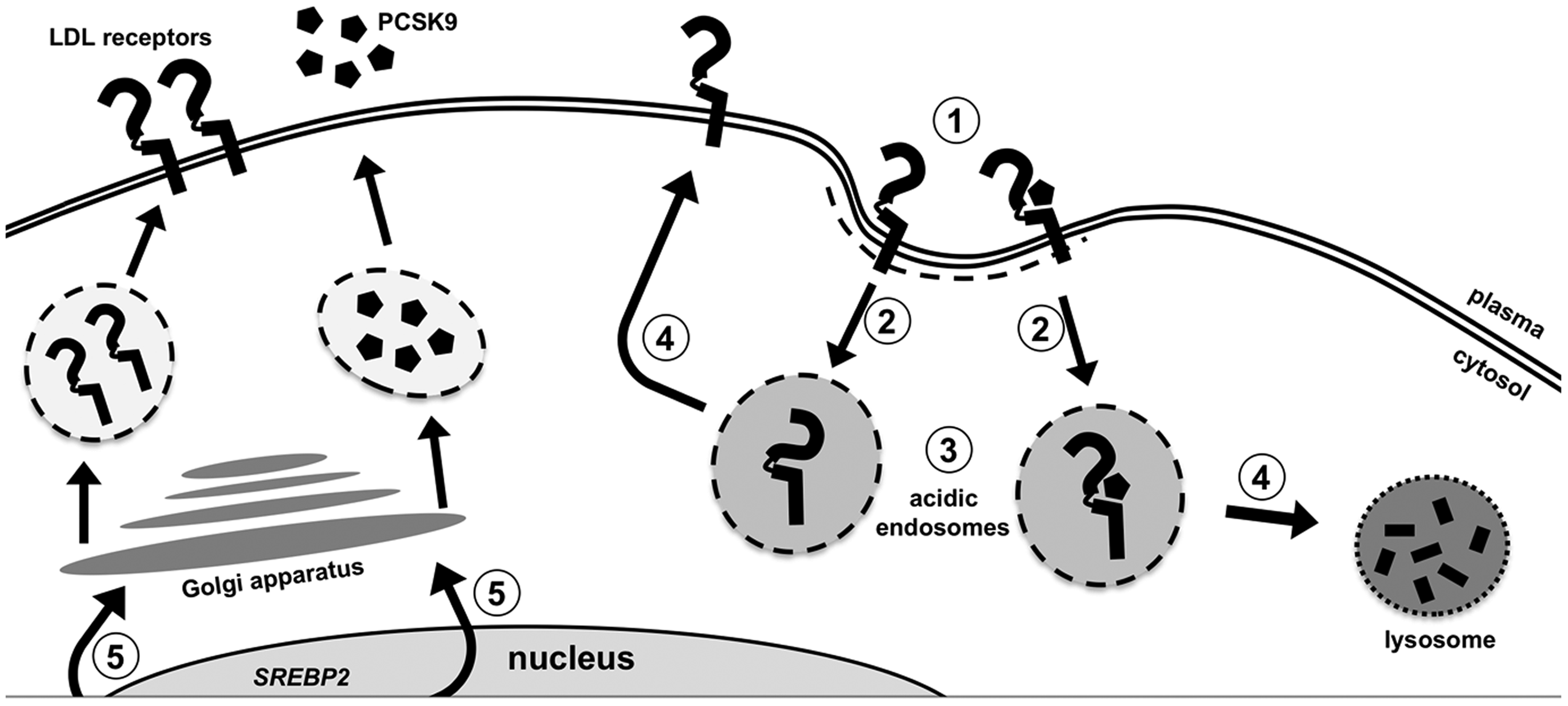
LDLR may be recycled up to 100 times during its lifetime. It is constantly being endocytosed, independent of LDL binding. LDLR life-cycle and role of PCSK9: (1) LDL receptor present on regions of cell membrane coated with Clathrin (aka ‘clathrin pits’); (2) Clathrin pits invaginate to form intracellular vesicles containing LDLR. This endocytosis is triggered by LDLRAP binding in hepatocytes (the cells responsible for the majority of LDL internalization); (3) Vesicles fuse with acidic early endosomes. The acidic environment causes conformational change which releases LDL. LDL is sorted to late endosomes and delivered to lysosomes where it is degraded; (4) The fate of LDL receptor is determined by the presence of PCSK9. If PCSK9 is absent, then LDLRs are recycled to the cell membrane. If bound to PCSK9, then LDLR and PCSK9 are targeted for lysosomal degradation; (5) Both PCSK9 and LDLR expression are turned on by SREBP2, which is activated by low concentrations of intracellular cholesterol. Recently synthesized LDLR can make its way to the cell membrane, or if bound to PCSK9 intracellularly will be targeted for lysosomal degradation.
In 2003, Abifadel et al. found that mutations in PCSK9 tended to co-segregate with severe hypercholesterolaemia in a number of French families. 10 Independent, but virtually simultaneous, molecular studies led by Seidah et al. in Canada revealed a new proprotein convertase, designated neural apoptosis-regulated convertase (NARC-1). 11 NARC-1 appeared to have a role in neurological development but Seidah et al. also found that NARC-1 was expressed in the liver, kidney and small intestine. They noted that NARC-1 occupied the same region of the human chromosome 1p32 identified by the French genetic studies of hypercholesterolaemia. This led them to speculate that NARC-1 might be implicated in FH. NARC-1 would later come to be re-designated as proprotein convertase subtilisin kexin 9, or PCSK9. Since 2003, a huge body of research ranging from molecular biology through to clinical genetic and epidemiological studies has characterized and established the key role that PCSK9 plays in regulating plasma LDL and thereby contributing to cardiovascular risk (CV risk). As this role emerged, a variety of pharmaceutical companies sought to determine whether PCSK9 might prove a suitable target for reducing plasma LDL. Perhaps the most notable resulting development was the demonstration in 2017 that inhibition of PCSK9 significantly reduces CV risk. This and other key developments3–17 in our current understanding of PCSK9, and FH, are summarized in Figure 2.
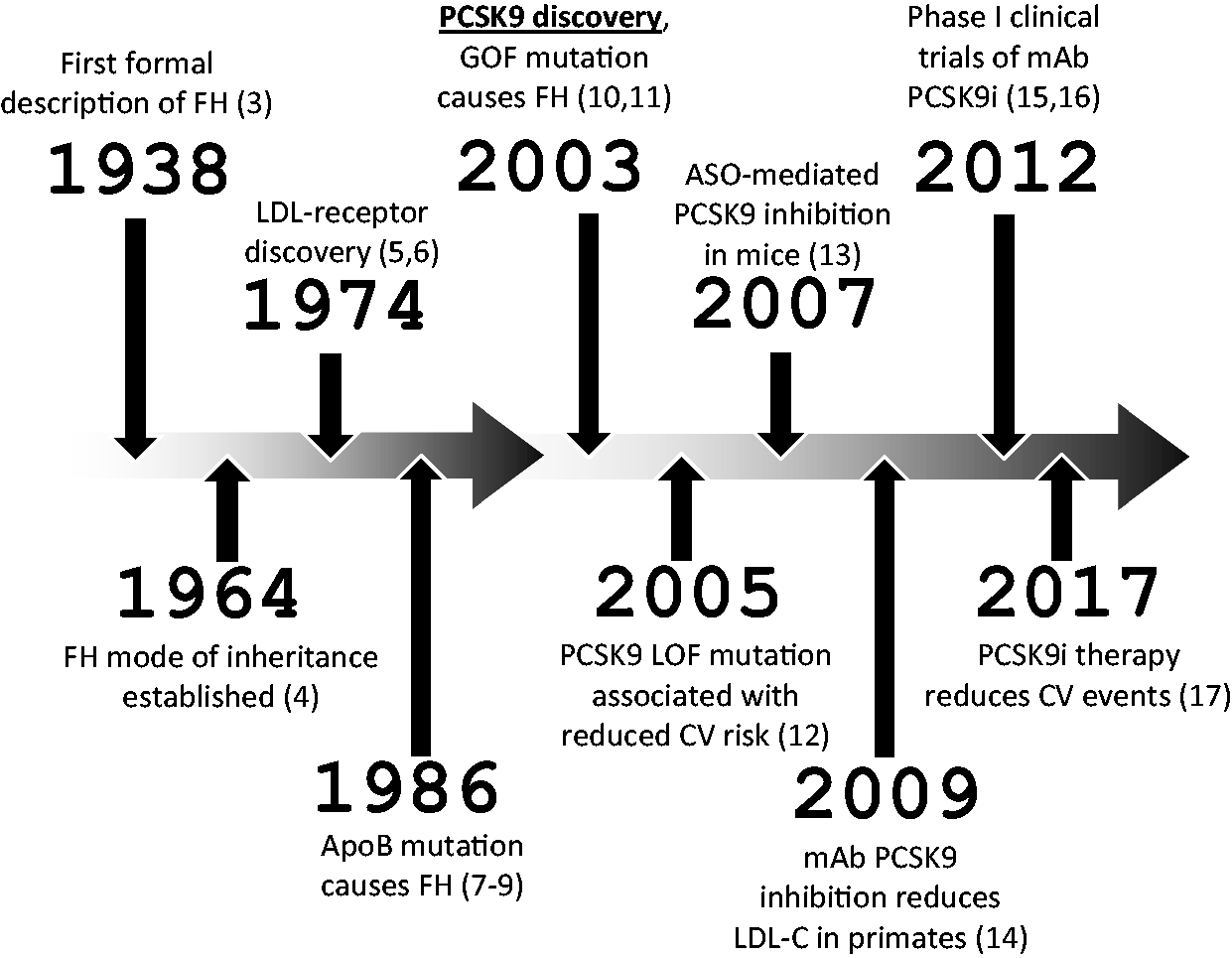
Key developments in the history of PCSK9 and FH.
PCSK9 role in LDL metabolism
Brown and Goldstein’s discovery of LDLR unveiled the principal mechanism by which LDL is removed from the circulation. LDLRs incorporated within the cell membrane of hepatocytes bind extra-cellular LDL and the resulting complexes are internalized. The full life-cycle of LDLR (see Figure 1 for a simplified schema) is complex: key steps involve expression, incorporation within the cell membrane, binding to LDL, internalization, then either recycling to the cell membrane or lysosomal degradation. The whole process is regulated by multiple proteins and remains incompletely understood. However, it is clear that PCSK9 has a key regulatory function as it determines the ultimate fate of LDLR: in the absence of PCSK9, internalized LDLR is recycled back to the cell membrane whereas PCSK9-bound LDLR is targeted for lysosomal degradation. Hence, in simple terms, increased PCSK9 chaperone activity reduces LDLR concentrations, leading to reduced LDL clearance and increasing plasma concentrations of LDL-C.
PCSK9 was first shown to reduce hepatocyte LDLR concentrations through manipulation of its expression in mouse models.18–20 It has been found to bind LDLR both extra- and intracellularly 21 although extracellular binding is probably the principal means by which LDLR concentrations are modulated. 22 PCSK9 binds LDLR in a 1:1 stoichiometry with an affinity that is dependent on calcium concentration, and which is markedly increased by lower pH. 23 The latter implies that the PCSK9-LDLR complex is much more tightly bound once internalized into acidic endosomes.
The means by which PCSK9 targets LDLR for degradation has been revealed by structural studies. 24 These show that PCSK9 binding changes LDLR’s structure from a closed to an open conformation. LDLR in a closed conformation is targeted for recycling to the cell membrane whereas PCSK9-bound LDLR, in an open configuration, is designated for lysosomal degradation.
A number of studies in humans have shown the expected positive correlation between plasma PCSK9 concentrations and LDL-C concentration.25–31 These are supported by animal studies where PCSK9 expression has been manipulated.18–20,22 Interestingly, PCSK9 concentrations can be influenced by a number of non-genetic factors, including drugs (most notably statins) and lifestyle, a phenomenon described in more detail in section ‘Measurement of, and influences upon, plasma PCSK9 concentrations’.
While concentrations of PCSK9 expression clearly influence LDLR activity (and therefore plasma LDL concentration), more dramatic effects can be exerted by alterations to the PCSK9 gene. Such mutations may result in altered levels of PCSK9 expression, or produce changes in the structure and therefore the activity of the PCSK9 protein. As described above, gain of function PCSK9 mutations were first discovered through French clinical genetic studies which identified PCSK9 as a cause of FH. 10 Conversely, loss of function (LOF) PCSK9 mutations in humans, leading to the opposite effect of reduced plasma LDL, have also been found.12,32–34 Many of the mutations observed in humans have been replicated in animal models and hepatocellular studies, a number of which have shed light on the role of particular functional domains within the PCSK9 protein. These and other studies of PCSK9 mutations are described in more detail in the following two sections.
Size, structure and LDLR-binding behaviour of PCSK9
The PCSK9 gene (also known as FH3, HCHOLA3 and NARC1) spans 3617 base pairs over 12 exons on chromosome 1p32. It is expressed primarily in the liver and intestine, but also within the kidney and nervous system, to produce a 692 amino-acid protein. This 75 kDa proenzyme comprises an N-terminal signal peptide followed by three principal domains: a prodomain, a catalytic domain and a C-terminal domain (CTD) (Figure 3).
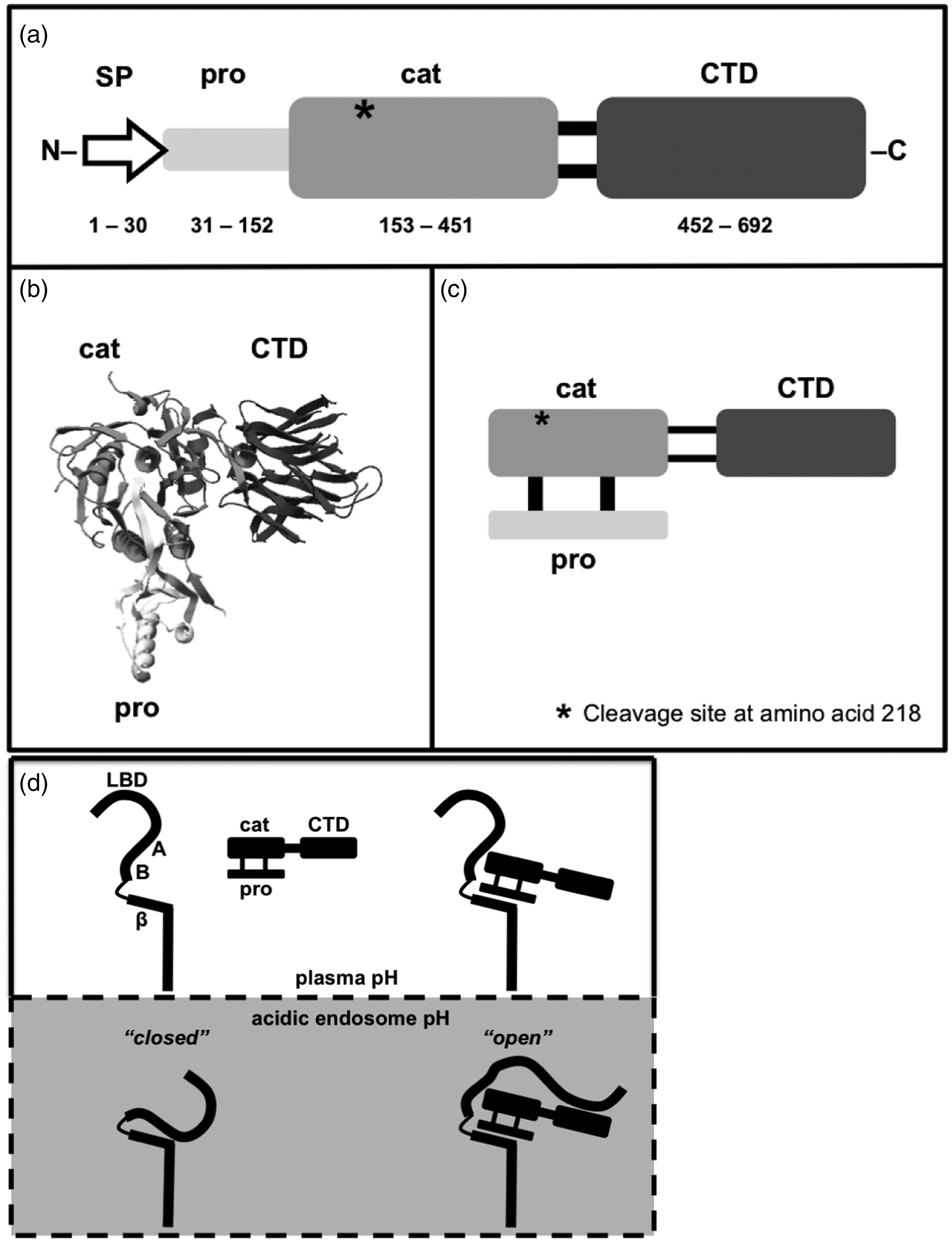
PCSK9 structure. (a) Functional domains and their corresponding amino acid ranges: signal peptide (SP), prodomain (pro), catalytic (cat), C-terminal domain (CTD). Autocatalytic cleavage results in the mature form of PCSK9 in which the prodomain binds non-covalently to the catalytic domain as represented by: (b) its crystal structure, (c) a schematic. Further cleavage of mature PCSK9 (c) at amino acid 218 (*) by furin or PC5/6A produces the inactive form of PCSK9. (d) A model of mature PCSK9 binding to LDLR: in the absence of PCSK9, LDLR adopts a ‘closed’ conformation within the acidic endosome. PCSK9 binding maintains an ‘open’ LDLR structure, targeting it for lysosomal destruction. Relevant LDLR domains: LBD: ligand-binding domain; A, B: epidermal growth factor-like repeats A and B; β: β-propeller region.
The short signal peptide targets PCSK9 for secretion from the cell but is cleaved from the rest of the protein to leave the three principal domains. Autocatalytic cleavage of the prodomain 35 is then required in order for the mature protein to be secreted from the endoplasmic reticulum (ER). 11 Structural studies23,36 have shown that the cleaved prodomain binds non-covalently to its previously neighbouring catalytic domain, and this binding precludes any further proteolytic activity. Thus while PCSK9 is a serine protease, its proteolytic action appears to be limited to the autocatalytic cleavage required for its secretion from the ER. Degradation of LDLR is therefore based on PCSK9’s ability to chaperone LDLR toward lysosomal destruction, rather than on any intrinsic proteolytic function.
The means by which PCSK9 targets LDLR for degradation has been revealed by a combination of crystallographic and binding studies. The nature of binding between the two proteins changes significantly from the extra-cellular neutral pH environment of the plasma, to that encountered post-internalization within the relatively acidic intracellular endosomes. At neutral pH, the crystallographic structure of the PCSK9-LDLR complex shows that both the prodomain and catalytic domain of PCSK9 are involved in LDLR-binding, while the CTD remains free (see Figure 3). The largest interaction by surface area is between the catalytic domain of PCSK9 and a region of LDLR referred to as EGF-A, where EGF-A is the first of three epidermal growth factor-like (EGF) repeats (the other two being EGF-B and EGF-C) located within the extra-cellular region of LDLR. A second smaller area of interaction occurs, through van der Waals contacts, between the prodomain of PCSK9 and the β-propeller region of LDLR.
There is no corresponding crystal structure for the PCSK9-LDLR complex at lower pH but there is a low pH structure for LDLR alone. 37 It reveals a ‘closed’ conformation (Figure 3) where a number of domains swing round in order to make intramolecular bonds with the β-propeller region. Comparison with the ‘open’ PCSK9-bound LDLR structure, reveals that PCSK9 binding would interrupt the contacts necessary to produce a ‘closed’ conformation. 24 It is therefore inferred that this structural difference, controlled by PCSK9, provides a means of determining whether an LDL receptor is destined for recycling to the cell surface, or degradation in the lysosomes.
The fate of PCSK9-bound LDLR is sealed by an increase in its affinity to PCSK9 at low pH. Studies show that the affinity of PCSK9 for LDLR increases by three to four-fold at pH 5,23 probably primarily because of the formation of salt bridges between the PCSK9 catalytic domain and the EGF-A region of LDLR.38,39 At acidic pH there also appears to be a contact formed between the CTD of PCSK9 and the ligand-binding domain (LBD) of LDLR.24,40,41 This is supported by binding studies at low pH which show dissociation of the complex in the absence of either the LBD or CTD, 41 and by hepatic cell assays where the LDLR-degrading capacity of PCSK9 is lost in a mutant lacking the CTD. 23
Two proprotein convertases (furin and PC5/6A) have been identified to inactivate mature PCSK9, via cleavage after amino acid 218 in the catalytic domain. 42 Interestingly, some individuals with hypercholesterolaemia have been found to have mutations near this site (see next section).10,43,44 These mutations inhibit cleavage of PCSK9 leading to increased concentrations of active PCSK9 and reduced LDLR concentrations. 44
Human PCSK9 mutations
PCSK9 mutations which cause a significant change in LDL-C concentrations can be divided into two groups, those which potentiate PCSK9 activity (‘gain of function’ mutations) and those which depress PCSK9 activity (‘LOF’ mutations). Gain of function (GOF) mutations lead to reduced LDLR concentrations, increased LDL-C in plasma and FH.10,43–47 LOF mutations, conversely, lead to elevated LDLR concentrations and reduced plasma concentration of LDL-C with evidence of protection against atheromatous cardiovascular disease (CVD).12,32–34 These findings raise some fundamental questions regarding the evolutionary benefit provided by PCSK9. It has been speculated that positive evolutionary pressures to conserve PCSK9 may stem from a reduced susceptibility to viral infection. This is based on in-vitro evidence that PCSK9 inhibits Hepatitis C virus entry into cells, 48 and on the hypothesis that a variety of viruses use LDLR to gain entry to cells. It could also be argued that increased PCSK9 activity may not have posed a significant risk for CVD in the preindustrial era, when diets were much less likely to have been high in saturated fat. Alternatively, a beneficial (but as yet uncharacterized) role for PCSK9 activity may lie in extra-hepatic tissues: rodent tissue and cell lines have demonstrated transient PCSK9 expression in the embryonic brain, and long-term expression in adult small intestine, kidney and pancreas. 11
Gain of function PCSK9 mutations which cause FH are relatively rare, probably responsible for around 3% of all monogenic hypercholesterolaemia, 49 while a much greater proportion of FH (60–80%) is accounted for by LDLR mutations. As noted in the introduction, a 2003 clinical genetics study of three French families with severe autosomal dominant hypercholesterolaemia lead to the discovery of the PCSK9 gene. 10 This ground-breaking work by Abifadel and co-workers revealed two separate PCSK9 GOF mutations: p.Ser127Arg which is located in the prodomain and p.Phe216Leu, located in the catalytic domain (see Figure 4).
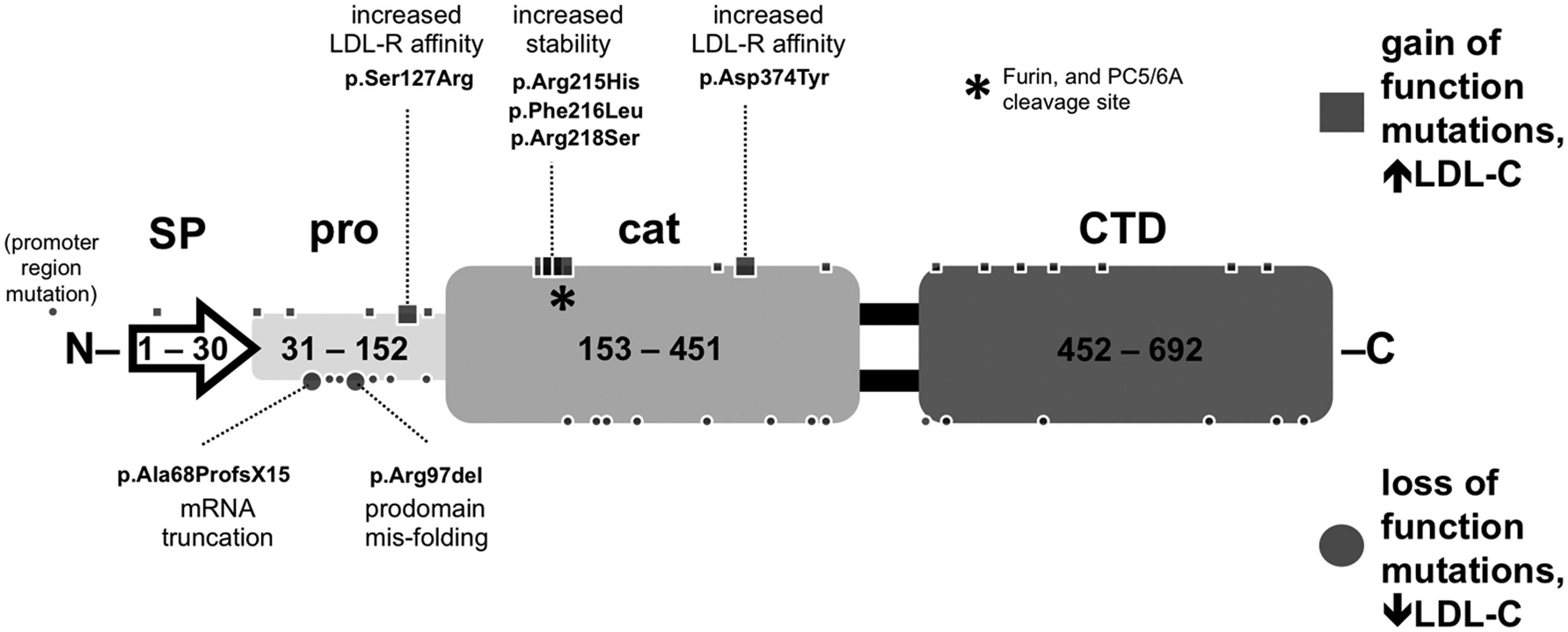
PCSK9 mutations identified in humans. Dots represent mutations identified in humans. Labelled mutations include those investigated by clinical genetics segregation studies and those where a mechanism of action has been identified. (Among the first gain-of-function mutations to be identified were those involving amino acids 215, 216 and 218. These appear to provide protection against furin or PC5/6A cleavage to the inactive form of PCSK9.)
An FH mutation database, hosted by University College London (http://www.ucl.ac.uk/ldlr/Current/), currently lists around 25 GOF PCSK9 mutations (see Figure 4 for a summary), including the original two mutations discovered by Abifadel et al. in 2003. Of these, the majority have only been described in single individuals. A total of five mutations have been shown to segregate with hypercholesterolaemia in family studies.10,43–47 In some cases, in-vitro cellular studies of the same mutations have been employed to demonstrate reduced LDL receptor concentrations.44,50–52 Structural and biophysical studies have provided potential mechanisms to explain the hypercholesterolaemia caused by specific mutations, including increased affinity for LDLR, 23 decreased PCSK9 degradation 23 and increased PCSK9 transcription. 53
LOF mutations are considerably more common than GOF mutations with prevalence of known mutations associated with hypocholesterolaemia having been found in the general population at around 2–3%.32–34,54 Of the approximately 21 known loss-of-function mutations (see Figure 4), only two have been shown to segregate within a family in an autosomal dominant fashion,55,56 the majority having been identified in unrelated individuals with hypocholesterolaemia.
LOF mutations have been shown to provide protection against atheromatous CVD and do not appear to be associated with any deleterious effects. 54 Interestingly, both animal 57 and human studies 58 suggest that LOF mutations confer an enhanced LDL-C lowering response to statin therapy. Statins lower intracellular cholesterol concentrations and therefore stimulate increased synthesis of LDLR via the sterol regulatory element binding protein (SREBP) pathway. This pathway also leads to the increased expression of PCSK9, whose targeting of LDLR for degradation would be expected to partially offset the benefit derived from increased LDLR expression. Hence, an individual with a PCSK9 LOF mutation on statin therapy might be expected to experience a smaller offset to their increased LDLR concentrations, and therefore an enhanced LDL-C lowering.
Three mutations have been shown to lead to deletions and subsequent truncation of PCSK9.55,56,59 The remaining LOF mutations are substitutions. In most cases the mechanism by which these substitutions produce LOF is unknown. Of those that have been characterized, substitutions have been found to impair PCSK9 secretion by impaired autocatalysis, or through abnormal folding.44,55
Measurement of, and influences upon, plasma PCSK9 concentrations
Measurement of plasma PCSK9 concentrations
Quantitation of total plasma PCSK9 is complicated by the cleavage of the mature form at residue 218 by furin, with production of an inactive product that is no longer a chaperone for LDLR destruction. 42 PCSK9 is also reported to show binding to apoB-containing lipoproteins. 60 PCSK9 measurement has generally been by ELISA immunoassay, but the majority of these assays have been unable to distinguish between the mature and cleaved forms. 61 There are however some ELISA methods which use monoclonal antibodies to distinguish the mature and cleaved forms. Using the latter technique in a population of homozygous FH (hoFH) patients undergoing LDL-apheresis, Hori et al. showed that both mature and furin-cleaved PCSK9 were both reduced by 55–56% after a single LDL-apheresis treatment. 62 In these patients, the furin-cleaved PCSK9 concentrations were 15% of the mature PCSK9 protein.
A novel method reported by Yeh et al. utilized the EGF-AB domain of LDLR to capture mature PCSK9 in a non-antibody ELISA method. 63 It is also possible to measure PCSK9 in serum using liquid chromatography-tandem mass spectrometry (LC-MS/MS). One such approach 64 involved trypsin proteolysis, followed by solid-phase extraction and subsequent LC-MS/MS identification of a peptide comprising 14 amino acids from the PCSK9 protein. This approach was found to correlate well with total plasma PCSK9 measured by the more usual ELISA methodology.
PCSK9 concentrations measured across different ELISA methods show a wide spread of results (40–800 ng/ml) and information on PCSK9 concentrations in clinical endpoint trials is very limited. 60 In general, concentrations appear to correlate with LDL-cholesterol and to increase on statin therapy (discussed in more detail in the section below ‘Effects of lipid-lowering drugs on plasma PCSK9 concentrations’).
At present, the wide variability of results significantly limits the utility of PCSK9 measurement in clinical practice. Even in the context of PCSK9 inhibiting therapy, the clinically relevant marker of efficacy remains the LDL-cholesterol value. Standardization, improved assay agreement and improved specificity for total vs biologically active PCSK9 might widen the scope of PCSK9 measurement but current clinical use, for example in assessing compliance with PCSK9 inhibitor medication, is limited.
Influences on plasma PCSK9
PCSK9 expression is principally modulated by intracellular cholesterol concentrations. In-vitro and animal studies have shown the link between intracellular cholesterol and PCSK9 activity to be mediated by a number of different transcription factors, 65 of which the most significant are SREBP257,66–70 and hepatocyte nuclear factor one-alpha (HNF-1α).70,71
Human plasma PCSK9 concentrations have correspondingly been found to correlate with plasma LDL-C and total cholesterol.25,29,30 Fasting leads to reduced PCSK9 expression and plasma concentrations,72–74 as does adhering to a diet high in polyunsaturated fats.75,76 When monitored throughout the day, PCSK9 concentrations have also been found to follow the diurnal rhythm of cholesterol. 74
Large studies, such as that involving an ethnically diverse patient group (n = 3138) derived from the Dallas Heart Study, have shown that the variation in PCSK9 serum concentration can be up to 100-fold. 29 In common with another similar study, 30 the distribution of PCSK9 values was right-skewed with a majority of individuals having relatively low concentrations and a minority with concentrations extending into a much higher range. A consistent finding is that women, particularly postmenopausal women, have significantly higher PCSK9 concentrations than men.29,30,77 While in men there does not appear to be a relationship between serum PCSK9 and age, in women PCSK9 is higher postmenopause and is inversely correlated with oestradiol. 77 PCSK9 has been found to be highest in the follicular versus mid-cycle or luteal phase 77 and significantly lowered in women undergoing IVF therapy, who experience supraphysiological concentrations of oestradiol. 78
Other variables which have been found to show a significant correlation with PCSK9 concentrations include fasting glucose, insulin, triglyceride concentrations29,30 and hepatic triglyceride content. 29 In keeping with these findings, PCSK9 concentrations are higher in those with obesity, or diabetes.29,30 However, the authors of the Dallas Heart derived PCSK9 study estimated that only 23% of the variation observed among participants could be explained by these and similar risk factors, and anticipated that only a very small proportion of the population would be affected by mutations leading to reduced PCSK9 expression. 29 Therefore, the very wide variation in serum PCSK9 concentration remains largely unexplained. A similarly sized but non-ethnically diverse Chinese study 30 found less although still significant variation in serum PCSK9, 10 to 20-fold versus the 100-fold observed in the Dallas study, 29 within its study population.
As outlined in the previous section, certain human PCSK9 mutations have been found to significantly alter PCSK9 expression levels. Gain of function (GOF) mutations in the promoter region of the PCSK9 gene have been shown to markedly increase PCSK9 expression, thereby resulting in hypercholesterolaemia. 53 A number of other GOF mutations reduce the rate by which active PCSK9 is cleaved into its truncated, non-active form by furin and PC5/6A proteolysis.10,43,44 Of the PCSK9 LOF mutations where a mechanism is known, many involve a reduction in PCSK9 serum concentration resulting from a failure to secrete PCSK9 from the cell.44,58,59,79 A further LOF mutation leads to reduced PCSK9 expression levels via truncation and degradation of its mRNA. 56
Effect of lipid-lowering drugs on plasma PCSK9 concentrations
Given that a number of lipid-lowering drugs (such as statins, fibrates and ezetimibe) exert their effects via increased LDLR expression mediated by SREBP-2, it might be expected that they would also increase PCSK9 expression. As the role that PCSK9 plays in LDLR turnover became clearer, it was hypothesized that the reduction in LDL-C achieved by such drugs might be partially offset by a concomitant increase in PCSK9 expression, and therefore increased LDLR degradation. 80
The first reports of a statin-mediated increase in PCSK9 concentrations involved individuals on atorvastatin.81,82 Subsequent studies utilizing simvastatin 83 and rosuvastatin 84 have shown similar results, and it would seem that the magnitude of the PCSK9 increase may be dose responsive. 85 However, analyses attempting to establish the relationship between the increase in PCSK9 concentrations and reduction in LDL-C that occurs with statin therapy have not produced consistent results. Some studies found that larger statin-mediated increases in PCSK9 translate to smaller LDL-C reductions,83,84 while others have found no significant correlation.81,86
Compared with statins, fewer studies have looked at the effect of ezetimibe on PCSK9 concentrations. It would appear from animal studies that ezetimibe alone is capable of quite significant increases in PCSK9,87 a finding consistent with two human studies which looked at the increase in PCSK9 caused by adding ezetimibe to existing statin therapy.31,88 However, not all studies have been able to replicate these results with non-significant results for both monotherapy86,89 and addition of ezetimibe to statin medication.83,89
The specific manner by which fibrates lower LDL-C remains largely unknown but appears likely to require the activation of multiple genes implicated in lipid metabolism, which are also known to alter VLDL and high-density lipoprotein (HDL) concentrations. The impact that a fibrate might be anticipated to have on PCSK9 concentrations is therefore less clear and has not been clarified by apparently conflicting results from experimental studies. Early cell-based studies showed that fibrates reduce PCSK9 expression in hepatocytes, 90 work corroborated by human studies which reported a reduced serum PCSK9 with fibrate therapy.26,91 However, numerous subsequent studies have instead reported increased PCSK9 concentrations.82,92–94
Lastly, a single session of LDL apheresis has been shown to reduce PCSK9 concentrations, by approximately 50%, in both heFH and hoFH patients participating in a small (n = 18) Japanese study. 62 In this study, both mature and furin-cleaved forms were reduced by similar proportions, and there was a high degree of correlation between the reduction in LDL-C and that of mature PCSK9. Immunoprecipitation studies using plasma from these patients showed binding between mature PCSK9 and ApoB. It was therefore inferred that a significant portion of the PCSK9 removed, does so while bound to ApoB-containing lipoproteins.
In summary, accumulating evidence indicates that statins increase serum PCSK9 concentrations, while studies on the effect of ezetimibe or fibrate therapy have produced conflicting results. Where a therapy does increase PCSK9 expression in tandem with LDLR, then serum PCSK9 concentrations will, to some extent, be dependent on the proportion of PCSK9 that becomes bound to LDLR and internalized. This aspect of PCSK9 behaviour may go some way to explaining some of the apparently conflicting results described above. Lastly, LDL apheresis has been shown to very effectively reduce serum PCSK9, and it seems likely that a significant proportion of the PCSK9 removed by apheresis does so while bound to ApoB-containing lipoproteins.
Drugs designed to specifically target PCSK9
As described in ‘Human PCSK9 mutations’ section, genetic studies looking at individuals and families with loss-of-function PCSK9 mutations have shown that their relative lack of PCSK9 activity confers significant benefit in terms of reduced LDL-C and CV risk without any known disadvantage. 54 The combination of these clinical findings with an emerging description of the mechanism by which PCSK9 regulates plasma LDL-C (from molecular, cellular and animal studies), led to PCSK9 inhibition being quickly identified as a novel means by which LDL-C might be safely lowered. The advent of PCSK9-inhibiting drugs promises a significant step forward for the prevention of CV disease in certain patient groups with hypercholesterolaemia. These include individuals for whom LDL-C remains unacceptably high despite maximal statin and ezetimibe therapy (and where plasmapheresis might be indicated), those in whom statin therapy is contraindicated or in those who find it difficult to tolerate conventional treatment. Some also see PCSK9 inhibitors as beneficial to a much wider patient population, as a means of achieving ever lower LDL-C and thereby ever lower CV risk.
A cornucopia of PCSK9 inhibitors
A large number of different PCSK9 inhibiting modalities have been investigated (see Table 1), although only a very small subset have so far made it beyond preclinical trials. The most successful to-date have been the monoclonal antibodies (mAbs), two of which (Alirocumab and Evolocumab) have completed phase 3 trials and are now recommended in the UK by NICE95,96 and SMC97,98 for individuals with FH or previous CVD, where LDL-C is judged to be refractory to maximally tolerated statin and ezetimibe therapy. In 2017, Evolocumab became the first PCSK9i to demonstrate reduced CV event rates, 17 followed by Alirocumab in 2018.99
PCSK9 inhibiting drug classes.
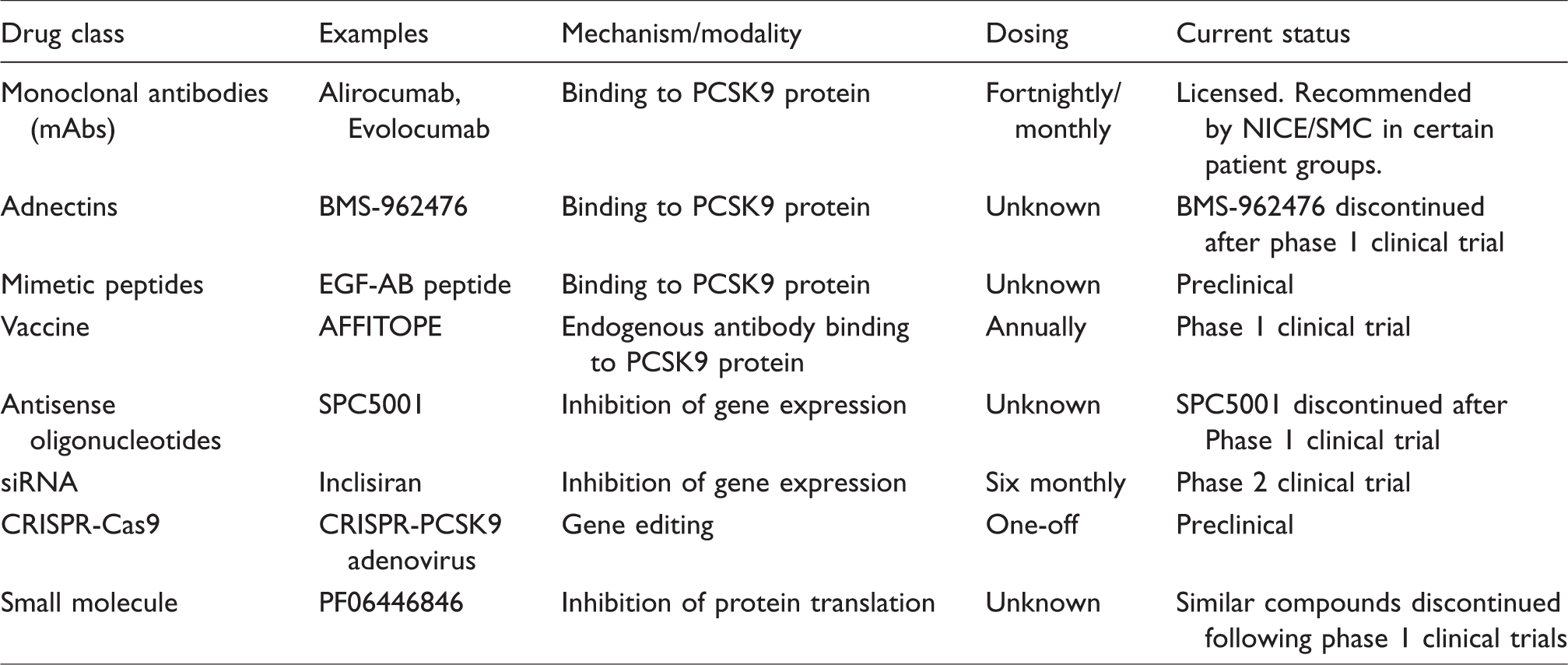
Although Alirocumab and Evolocumab are currently the most high-profile PCSK9 inhibitors, the first forms of PCSK9 inhibition to be investigated involved quite a different mode of action. In 2007, preclinical studies showed that PCSK9 expression could be successfully suppressed using antisense oligonucleotides (ASO). 13 Similar evidence followed slightly later for small interfering RNA (siRNA). 100 However, despite the early success of ASO-mediated inhibition, a number of subsequent phase 1 trials were terminated early, several without explanation. One of the trials (for an ASO preparation named SPC5001) that did report outcomes described adverse effects in the form of injection-site reactions and nephrotoxicity. 101 In contrast, siRNA approaches have fared much better, and indeed, a recent phase 2 trial of subcutaneously delivered Inclisiran has demonstrated an efficacy in LDL-C reduction which approaches that of the most successful mAb treatments. 102 A phase 3 study of Inclisiran (ORION-4) is anticipated to start in early 2019, and it is to be trialled with a dosing frequency (dose at baseline, 90 days, and thereafter six monthly) that compares very favourably with the fortnightly or four-weekly regimens of the mAbs.
A more definitive approach to manipulating PCSK9 expression is that promised by CRISPR-Cas9, a gene-editing technology adapted from a mechanism by which bacteria rid themselves of viral DNA. Musurunu et al. have shown that adenovirus delivery of a CRISPR-Cas9 system is able to efficiently disrupt PCSK9 expression in mice, resulting in permanently reduced serum concentrations of LDL-C. 103 If this approach could be translated to humans, it would provide a revolutionary one-off treatment for chronic hypercholesterolaemia. However, with a technique that involves making permanent genetic changes, a substantial amount of work remains to be done to demonstrate its safety, the most obvious concern being that of possible off-target gene editing. Hence, the balance of risk versus benefit for CRISPR may be judged more appropriate for patients with hoFH, where severe or null LDLR mutations make the provision of functional LDLR expression a live-saving intervention and could spare the need for liver transplant and long-term immuno-suppressive therapy.
In a bid to produce a small molecule inhibitor of PCSK9 with the potential for oral administration, Pfizer developed PF06446846. This compound was the end result of a phenotypic screening assay, which assessed the ability to inhibit PCSK9. A subsequent collaboration with researchers at the University of California Berkeley uncovered the mechanism by which PF06446846 achieves its inhibition. 104 It was found that, by binding to the ribosome and its emerging amino acid chain, this small molecule is capable of blocking protein synthesis with unprecedented specificity, in this case only doing so for PCSK9 and approximately 20 other proteins. However, despite its initial promise, Pfizer has discontinued the development of PF06446846 and two distinct but similar compounds.
Another class of PCSK9 inhibitor includes inhibitory proteins or peptides, which like mAbs, target the PCSK9 protein rather than its expression. Two types have been developed: mimetic peptides and adnectins (or monobodies). The mimetic peptides (which have not, as yet, progressed beyond preclinical trials) bind to PCSK9 by mimicking the binding domains of LDLR, and thereby competitively inhibit PCSK9’s interaction with full-length LDLR. 105 The adnectins are much larger synthetic binding proteins whose common structure is based on a domain of fibronectin. These have shown promise in animal studies 106 but have not progressed beyond a phase 1 trial. 107
A further means of inhibiting the PCSK9 protein comes from a rather unexpected quarter. Two independent groups of researchers have successfully completed initial steps toward a PCSK9 vaccine. This pioneering work aims to harness the immune system in order to produce endogenous PCSK9 inhibiting antibodies. Affiris has taken the development of its vaccine (AFFITOPE) from promising animal studies 108 through to a phase 1 clinical trial, 109 while an academic group based at NIH Bethesda have demonstrated efficacy in mice and macaques. 110 It is anticipated that a successful vaccine will require an annual booster in order to maintain antibody titres sufficient to effectively inhibit PCSK9. However, this would still represent a potentially significant advantage over the fortnightly (or four-weekly) dosing necessary with mAbs.
Development of the monoclonal antibody PCSK9 inhibitors
In 2009, preclinical work demonstrated the potential efficacy of mAb-mediated inhibition with cellular studies that showed increased LDLR concentrations upon exposure to inhibiting antibodies. 111 This was swiftly followed by animal studies demonstrating reduced LDL-C in mice and primates given a PCSK9-inhibiting monoclonal antibody. 14 This latter work also involved in vitro analysis, which showed that a combination of PCSK9 inhibitor and statin exerted a synergistic effect upon LDLR concentrations.
These promising results led to the rapid development of mAb preparations by multiple pharmaceutical companies, including Amgen (Evolocumab), Eli Lilly (LY3015014), Novartis (LGT-209), Merck (1D05-IgG2, 1B20), Roche (RG7652), Sanofi/Regeneron (Alirocumab) and Pfizer (Bococizumab, J10, J16, J17). Of these, many mAbs have made it beyond preclinical studies, but only a few have progressed beyond phase 2 clinical trials (Alirocumab, Bococizumab and Evolocumab), while LGT209 has interestingly been relicensed to Cyon Therapeutics for the purpose of a phase 2 study focusing on the potential beneficial effects of PCSK9-inhibition in patients with sepsis. 112
Both Alirocumab and Evolocumab are fully human mAbs, while Bococizumab is a humanized mAb that retains some murine sequences within the variable portion of the antibody. Bococizumab was discontinued after phase 3 clinical trials showed variable LDL-C reduction. This phenomenon seems to have been caused by the development of anti-Bococizumab antibodies in a large proportion of patients 113 and may reflect the inclusion of potentially antigenic murine sequences.
In contrast, Alirocumab and Evolocumab have demonstrated consistent and long-term LDL-C lowering effects114,115 and are now both licensed in the USA and EU. Assessments of each drug, conducted by NICE95,96 and SMC,97,98 have recommended both as an option in the UK NHS for specific patient groups (see Table 2) with primary hypercholesterolaemia (or mixed dyslipidaemia), where existing maximally tolerated lipid-lowering therapy has failed to consistently reduce LDL-C below specific thresholds. However, funding restrictions at a local level, and the relatively high cost of Alirocumab and Evolocumab, have meant that wholescale adoption of the NICE/SMC recommendations has been quite variable across the UK. The lack of a trial demonstrating a reduced rate of CV events as the primary endpoint, until the recent FOURIER trial for Evolocumab in 201717 and ODYSSEY OUTCOMES for Alirocumab in 2018,99 may also have been a factor in this variable uptake.
NICE/SMC eligibility criteria for Alirocumab/Evolocumab therapy.
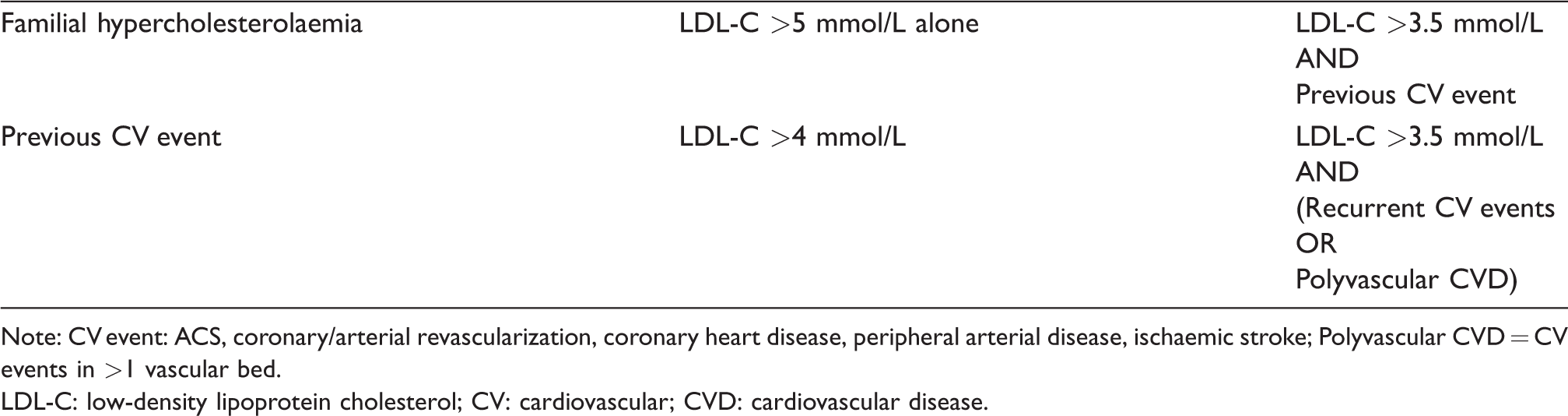
Note: CV event: ACS, coronary/arterial revascularization, coronary heart disease, peripheral arterial disease, ischaemic stroke; Polyvascular CVD = CV events in >1 vascular bed.
LDL-C: low-density lipoprotein cholesterol; CV: cardiovascular; CVD: cardiovascular disease.
Alirocumab and Evolocumab
Both Alirocumab (Praluent) and Evolocumab (Repatha) are human IgG monoclonal antibodies, produced in Chinese hamster ovary cells by recombinant DNA technology. They are administered by subcutaneous injection using a single use prefilled pen and until required are stored at 4°C. Standard doses can be delivered every two weeks (Q2W), or every four weeks (Q4W) where the clinically equivalent four-weekly dose is higher than the two-weekly dose. Both are indicated in adults with primary hypercholesterolaemia or mixed dyslipidaemia. Both can also be administered at higher doses. Alirocumab is available at 150 mg (double the standard dose of 75 mg) at a two-weekly frequency for patients requiring a larger LDL-C reduction. And Evolocumab can be prescribed at 420 mg (triple the standard dose of 140 mg) on a two-weekly basis in hoFH. In this latter group, the age range in which Evolocumab is indicated extends down to ≥12 years old. No dose adjustment is required for weight, or mild to moderate renal or liver impairment.
Alirocumab and Evolocumab: Adverse effects and safety
Long-term trials have demonstrated both Alirocumab (78 weeks) 114 and Evolocumab (52 weeks) 115 to be safe and well-tolerated, with similar proportions of patients reporting adverse effects in both treatment and non-treatment groups. Adverse effects leading to drug discontinuation in these trials were 7.2% (vs. 5.8% for placebo, P-value 0.26) for Alirocumab, and 2.4% for Evolocumab (no placebo group). Common side-effects, with a designated frequency of between 1:100 and 1:10, for Alirocumab were: oropharyngeal pain, rhinorrhoea, sneezing, pruritus and injection-site reactions (including local erythema, itching, swelling and tenderness). 116 Common side-effects reported for Evolocumab were similar in nature: nasopharyngitis, upper respiratory tract infection, influenza, rash, nausea, back pain, arthralgia and injection-site reactions (including erythema, pain and bruising). 117 Rare instances, frequency 1:10,000 to 1:1000, of hypersensitivity have been reported for both drugs.114,118 Encouragingly, a recently reported observational extension to the OSLER-1 study of Evolocumab did not reveal any overt increase in adverse effects, nor any significant loss in LDL-C lowering efficacy, over an average period of almost four years. 119
The development of antidrug antibodies, which may mediate either adverse effects or impair efficacy, is a potential concern with antibody-based drugs such as Alirocumab or Evolocumab. The measurement of antidrug antibodies has not been reported for all of the phase 2 and 3 clinical trials of these mAbs. However, it would appear that where measured, rates have always been low. For Evolocumab, only 0.1% of patients screened (n = 4846) tested positive for anti-Evolocumab antibodies, and none were found to have neutralizing antibodies. 117 For Alirocumab, 4.8% of patients were determined to have antidrug antibodies, and 0.3% were found to be positive for neutralizing antibodies on ≥2 occasions. 116 Where neutralizing antibodies to Alirocumab were detected, there does not appear to have been an effect on LDL-C lowering efficacy. 116
Safety and efficacy data are relatively limited for patients ≥75 years, with only just over 200 patients represented in clinical trials for each of Alirocumab and Evolocumab. However, there do not appear to be any significant differences in safety or efficacy when compared to younger patients. Data for children are even further limited. No data are available for Alirocumab below the age of 18 years, although a clinical trial called ODYSSEY KIDS for ages 8–17 years is currently underway. 120 Evolocumab has been trialled in a small number (n = 14) of hoFH individuals ranging in age from 12 to 18 years, and a larger phase 3 trial called HAUSER-RCT for ages 10–17 years is ongoing. 121
Theoretical concerns have been raised regarding the safety of sustaining low plasma LDL-C over long periods of time. It has been hypothesized that reduced availability of cholesterol (via LDL-C) to peripheral tissues might result in problems with steroidogenesis, fat-soluble vitamin transport and neurological dysfunction. Reassuringly, neurocognitive problems have not been reported in individuals with naturally occurring PCSK9 loss-of-function mutations, and therefore life-long low plasma LDL-C. 54 However, although such individuals maintain a lower than average LDL-C (∼3 mmol/L), 54 this concentration remains much higher than the average LDL-C achieved in PCSK9i clinical trials (∼1.5 mmol/L).114,115 These trials were not designed to formally measure neurocognitive effects, although both did report a higher rate of neurocognitive events in their treatment groups: Alirocumab (1.2% vs. 0.5%, P = 0.17) 114 and Evolocumab (0.9% vs. 0.3%). 115 Further analysis of these results has not revealed a correlation between lower LDL-C and a higher rate of neurocognitive events.115,122 Nor were there any clinically significant effects on cortisol, reproductive hormones, or fat-soluble vitamin concentrations. 122 A more recently reported RCT (EBBINGHAUS, n = 1204), employing formal cognitive assessments, has also failed to detect any significant increase in neurocognitive events for treatment with Evolocumab. 123 There remains a possibility of such side-effects occurring in the longer term, and a five-year extension 124 to this latter study (of median length 19 months) is ongoing.
Alirocumab and Evolocumab: Effect on LDL-C and other lipid fractions
Two large multistudy programmes (ODYSSEY for Alirocumab, PROFICIO for Evolocumab), each incorporating in excess of 20 clinical trials, have established that both Alirocumab and Evolocumab produce significant reductions in LDL-C. Using a Q2W frequency, a near steady-state LDL-C concentration is achieved after three doses of each drug, approximately five weeks from baseline. These LDL-C reductions are maintained into the long-term with open-label trials demonstrating an average fall of ∼60% for both drugs at 24 weeks (see Table 3) for a mixture of patients, including those with heFH, non-FH and statin intolerance.114,115 In these studies, this corresponded to a baseline LDL-C of ∼3.2 mmol/L falling upon treatment to ∼1.3 mmol/L. Significant reductions were also seen in non-high-density lipoprotein cholesterol (HDL-C) (∼50%) and triglycerides (∼15%), while there were small positive adjustments to HDL-C.
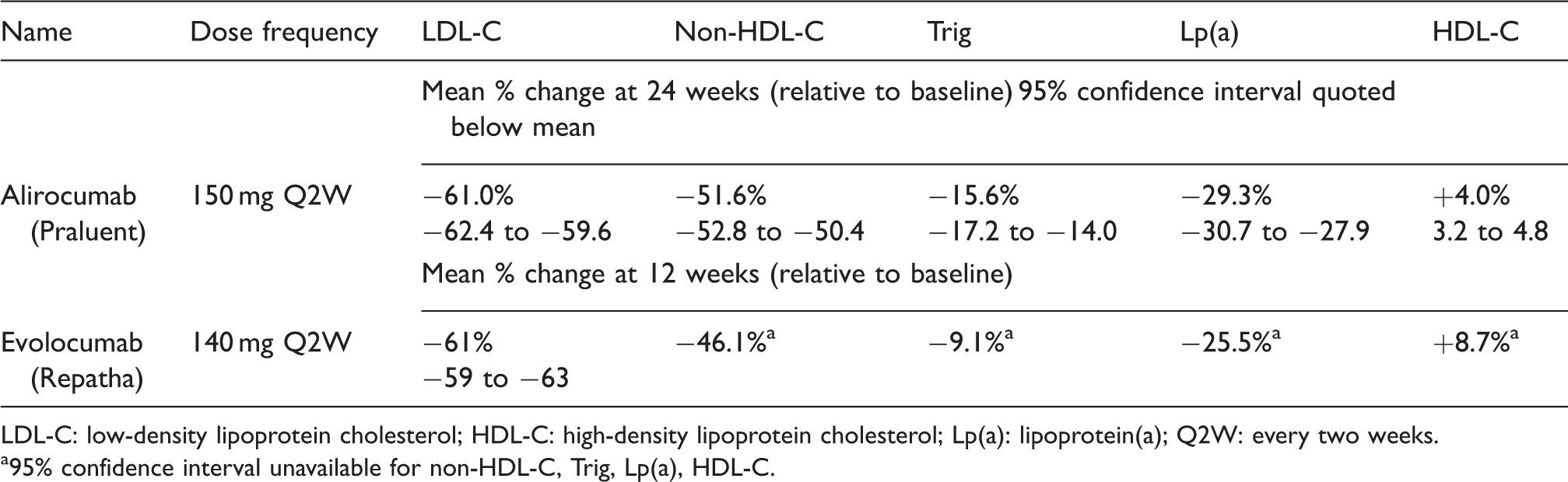
LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol; Lp(a): lipoprotein(a); Q2W: every two weeks.
95% confidence interval unavailable for non-HDL-C, Trig, Lp(a), HDL-C.
Unlike statin therapy, mAb-mediated PCSK9 inhibition also provides a significant reduction in lipoprotein(a) (Lp(a)), amounting to a fall of between 25 and 30%.114,115 While Lp(a) metabolism may involve several different mechanisms, it would therefore seem that the LDL receptor must play a significant role in removing Lp(a) from plasma in the context of PCSK9 inhibition. This is indicated by an analysis of clinical trials revealing a correlation between Lp(a) reduction and LDL-C reduction,125,126 and a number of in vitro125,127 and in vivo studies 125 demonstrating enhanced Lp(a) uptake for low concentrations of LDL-C and up-regulation of LDLR.
The anticipated synergistic effect of combining PCSK9i therapy with statin treatment (through off-setting statin-induced increased PCSK9 expression) has not been consistently demonstrated in clinical studies to date. Instead, subgroup analyses have generally shown similar percentage LDL-C reductions for those on Alirocumab or Evolocumab monotherapy, compared with those on a combination of statin and PCSK9i.128–130 There were also no significant differences in percentage LDL-C reduction with Alirocumab or Evolocumab between those established on regimens of varying different statin intensities.128,131 In contrast, the ODYSSEY CHOICE II study 132 did find slightly higher percentage LDL-C reductions with Alirocumab for those already established on a statin compared with those not receiving statin treatment.
A comparison of individuals with heFH, versus non-FH, does not seem to alter LDL-C response to PCSK9i-mAb therapy. 15 However, unsurprisingly, hoFH patients with two LDLR mutations do see much more modest LDL-C reductions, ∼20%. 133
Alirocumab and Evolocumab: Efficacy in FH
Of the various patient groups studied, the results for those with FH were among the most eagerly awaited by lipidologists. Individuals with FH have a particularly high CV risk, conferred by a lifetime of untreated or suboptimally controlled hypercholesterolaemia. Many patients see their CV risk reduced to levels comparable with the general population through high-intensity statin therapy. 2 However, a subset fails to achieve LDL-C targets, despite maximal oral therapy and may require plasmapheresis which is expensive, invasive and time consuming. Hence, it was hoped that PCSK9 inhibitors might provide a significant unmet need.
Fortunately, both Alirocumab and Evolocumab have been shown to produce significant further LDL-C reductions in FH patients already established on maximal oral therapy. HeFH patients treated with Alirocumab 134 achieved an average LDL-C reduction of ∼49% (95% CI: 46% to 52%) from baseline, a drop from mean LDL-C of ∼3.6 mmol/L to 1.8 mmol/L, and this was maintained until 78 weeks. A similar percentage LDL-C reduction was obtained in a smaller heFH study 135 where the baseline LDL-C was significantly higher at ∼5.1 mmol/L, achieving an average endpoint LDL-C of 2.8 mmol/L. RUTHERFORD-2 (n = 331) showed that heFH patients treated with Evolocumab also achieved significant LDL-C reductions of ∼60% (95% CI: 58% to 65%) from an average baseline of ∼4.2 mmol/L (to ∼1.7 mmol/L) over a period of 12 weeks. 136 A subsequent open-label extension to RUTHERFORD-2 has shown that these reductions are broadly maintained up to 48 weeks. 137
Further studies relevant to FH include ODYSSEY ESCAPE, 138 a clinical trial whose aim was to evaluate whether the frequency of lipoprotein apheresis might be reduced in the context of Alirocumab therapy, and a series of trials133,139 involving the treatment of hoFH patients with Evolocumab. Encouragingly, ODYSSEY ESCAPE (n = 62) reported that the average preapheresis LDL-C fell by about 43% (95% CI: 33% to 52%) which meant that 63% of the heFH patients (n = 41) achieved the trial’s treatment goal (of an LDL-C reduction of ≥30%) and were able to discontinue apheresis altogether. Interim results from the TAUSSIG study 133 looking into the safety and efficacy of Evolocumab for hoFH patients (n = 106, both receiving, and not receiving apheresis) have also been encouraging. After 12 weeks, a mean LDL-C reduction of ∼20% was achieved, and this was sustained at 48 weeks. Additionally, a higher dose (three times the regular two-weekly dose) of 420 mg was found to be well tolerated and to produce an additional ∼8% reduction relative to baseline LDL-C for a subset of patients (n = 47). The characterization of FH genes in TAUSSIG enabled researchers to characterize the relationship between genetic status and response to PCSK9 inhibition. Unsurprisingly, those with null LDLR mutations saw little benefit from therapy, whereas those with two mildly defective LDLR mutations and those with a combination of LDLR and either PCSK9 or ApoB mutations experienced the largest LDL-C reductions. Interestingly, Lp(a) reductions were much smaller than those seen in the heFH studies, again suggesting that its clearance from plasma is at least partly LDLR-mediated.
Alirocumab and Evolocumab: impact on plaque regression and CV events
In 2016, a further Evolocumab study reported the effects of PCSK9 inhibition on coronary artery imaging. GLAGOV or ‘Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound’ followed individuals with known angiographic coronary artery disease over 78 weeks. 140 Participants were randomized to receive either Evolocumab and statin therapy, or statin therapy alone. Percentage change in atheroma volume from baseline to week 78 was found to be significantly decreased in the Evolocumab group, and the proportion of individuals experiencing plaque regression was also significantly higher in the Evolocumab-treated group (64% vs. 47%). Patients in the treatment group achieved a mean LDL-C concentration of 0.95 mmol/L, and it remains an open question as to whether such low LDL-C concentrations might be expected to yield a higher proportion of patients with plaque regression. Perhaps, the process of plaque regression in some individuals requires more than 78 weeks, or perhaps there are other pathophysiological factors which limit the plaque-reducing efficacy of LDL-C lowering therapy.
A very significant milestone in the assessment of PCSK9 inhibition was reached in the first half of 2017. FOURIER, a study of Evolocumab efficacy, was the first PCSK9 inhibitor trial to report CV events as its primary endpoint. 17 FOURIER recruited patients with a background of atherosclerosis and demonstrated that the addition of Evolocumab to a lipid-lowering regimen, compared with statin treatment alone, conferred a significantly reduced risk for both the primary (cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina and coronary revascularization) and secondary (cardiovascular death, myocardial infarction and stroke) composite endpoints. However, FOURIER did not demonstrate a significant effect on the risk of CV death, or all-cause mortality.
A corresponding CV endpoint study for Alirocumab (ODYSSEY OUTCOMES)99,141 was published in the autumn of 2018. ODYSSEY OUTCOMES recruited patients with a history of acute coronary syndrome that had occurred between 1 and 12 months earlier and despite maximally tolerated statin therapy maintained an LDL-C of ≥1.8 mmol/L (or similarly elevated non-HDL-C, or ApoB). Like FOURIER, it demonstrated a significantly reduced risk for its primary composite endpoint (of death from CHD, non-fatal myocardial infarction, fatal or non-fatal ischaemic stroke, or unstable angina requiring hospitalization). There were multiple secondary endpoints, including any CHD event, any cardiovascular event and correspondingly death from CHD, death from CVD and all-cause mortality. And in contrast to FOURIER, there was a statistically significant reduction in all-cause mortality (hazard ratio, 0.85; 95% CI: 0.73 to 0.98; P = 0.026). However, as in FOURIER, neither the risk of mortality from CHD (or CVD) was significantly different between the placebo and PCSK9i arms.
Both FOURIER and ODYSSEY OUTCOMES were very large RCTs (n = 27,564 and 18,924 respectively), which recruited patients aged ≥40 years, with FOURIER also excluding those aged over 85. The study populations shared a number of characteristics, in that they were predominantly white (80 to 85%), male (75%) and of similar mean age (59 years in ODYSSEY OUTCOMES, 63 years in FOURIER). However, they did differ significantly in at least two respects: the nature of CV disease used as inclusion criteria, and consequently, the proportion of patients who had been established on long-term statin therapy. Those recruited to FOURIER qualified on the basis of having a range of different atherosclerotic diseases, including myocardial infarction (80%), non-haemorrhagic stroke (19%) or symptomatic peripheral artery disease (13%). (Inclusion criteria also required a specific background of high CV-risk characteristics.) In contrast, ODYSSEY OUTCOMES picked participants with a recent history of ACS and consequently included a much higher proportion of patients who had only relatively recently started high-intensity statin therapy.
The trials also differed in their approach to target LDL-C, and therefore in the long-term, LDL-C concentrations achieved. ODYSSEY OUTCOMES aimed to bring Alirocumab-treated patients down to an LDL-C range of between 0.6 and 1.3 mmol/L. (This was achieved by a protocol which increased the 75 mg dose to 150 mg in those with LDL-C > 1.3 mmol/L, and switched those achieving LDL-C < 0.4 mmol/L to placebo.) FOURIER did not attempt to limit very low LDL-C, and Evolocumab has a single dose concentration of 140 mg fortnightly (or 420 mg monthly). Hence, although median baseline LDL-C was 2.4 mmol/L in both trials, FOURIER saw falls to 0.8 mmol/L at two years, compared with 1.5 mmol/L in ODYSSEY OUTCOMES. Some of the difference in long-term LDL-C concentrations between the two studies can be explained by the use of the ODYSSEY OUTCOMES protocol which prevented sustained LDL-C < 0.4 mmol/L. This would also seem to be supported by the LDL-C results from ODYSSEY LONG TERM, 114 which did not employ such a protocol. ODYSSEY LONG TERM used a single dose concentration of Alirocumab 150 mg fortnightly and showed both similar percentage LDL-C reductions to Evolocumab 115 and a similarly sustained reduction in long-term LDL-C (see Table 3).
Having demonstrated considerable LDL-C reductions, FOURIER and ODYSSEY OUTCOMES reported corresponding statistically significant reductions, in both cases of 15%, in the primary composite endpoints (see Table 4) between placebo and treatment. For FOURIER, this involved following a population of patients with atherosclerotic disease over a median follow-up of 2.2 years. While for ODYSSEY OUTCOMES, recruited patients had a recent history of ACS and were followed up at a median of 2.8 years. Hence, although recruiting rather different groups of patients and involving different lengths of follow-up, both trials reported fairly similar absolute reductions in risk (ARR) and numbers needed to treat (NNT): for FOURIER, these were ARR 1.5%, NNT 67, and for ODYSSEY OUTCOMES, ARR 1.6%, NNT 63. FOURIER did not find a statistically significant reduction in CV mortality or all-cause mortality. In contrast, ODYSSEY OUTCOMES did detect lower all-cause mortality but, perhaps surprisingly, there was no statistically significant effect on CV mortality.
Cardiovascular endpoint results for treatment with a PCSK9i.
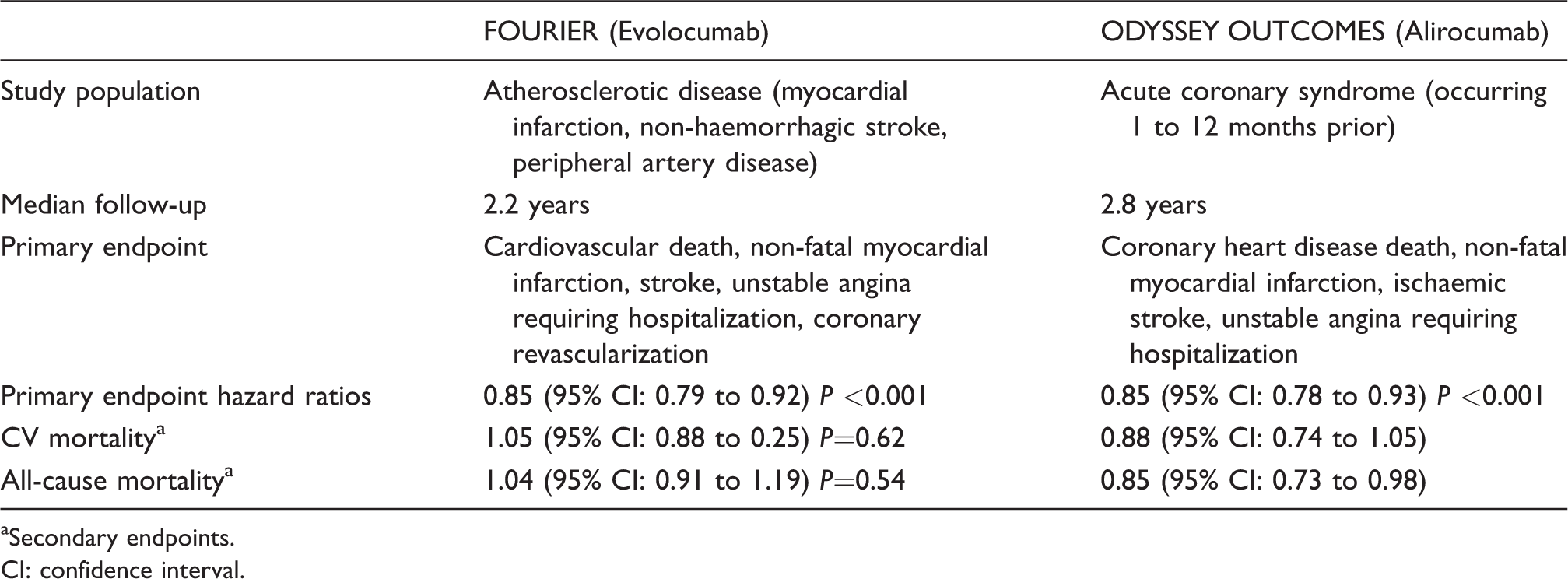
Secondary endpoints.
CI: confidence interval.
Given the very large percentage reductions in LDL-C produced by both Evolocumab and Alirocumab, it is probably safe to say that many had expected (rather than hoped) that their CV endpoint trials would provide a convincing demonstration of reduced CV event rates. Hence, the rather modest reduction in absolute risk, NNT of ∼65 and the absence of a statistically significant effect on CV mortality may have been disappointing.
However, the results are perhaps not so unsurprising given the relatively low baseline LDL-C (median 2.4 mmol/L) of those recruited. Previous high-intensity statin and statin–ezetimibe combination trials, 142 where the baseline LDL-C has also been relatively low, have similarly failed to demonstrate an effect on CV mortality. In fact, the FOURIER trialists assert that the CV risk reduction provided by Evolocumab is no less impressive than that of statins. Given the same absolute reduction in LDL-C over the same time interval, they point to very similar reductions in CV endpoint rates for statin therapy,143,144 an assertion which implies that the CV risk reduction is almost entirely attributed to LDL-C lowering effects. The FOURIER authors also suggest that the difference in CV risk between placebo and Evolocumab groups may widen with time, although this obviously remains somewhat speculative.
Perhaps in response to perceived disappointment in FOURIER, analyses stratifying individuals by baseline LDL-C were invoked in ODYSSEY OUTCOMES. For those with a baseline LDL-C of >2.6 mmol/L, the ARR was 3.4% (corresponding to NNT of 29) over the 2.8 years of the trial. Calculations projecting benefit over a longer period of four years, then estimated a much more compelling NNT of 16 (95% CI: 11 to 34).
In the UK, budgetary constraints on healthcare spending mean that very widespread prescribing of agents such as Alirocumab and Evolocumab is unlikely to occur in the near future. Advisory bodies such as NICE and the SMC have issued guidance95–98 recommending PCSK9i therapy for use by specialist clinics in those with threshold LDL-C concentrations significantly higher than the baseline median LDL-C of 2.4 mmol/L in FOURIER and ODYSSEY OUTCOMES. And in fact, it is our experience that many centres in the UK are adopting rather more restrictive criteria than those recommended by NICE/SMC. Precisely what threshold LDL-C and associated CV risk background should qualify an individual for PCSK9i therapy may prove rather hard to define. However, it seems quite reasonable to expect much larger reductions in absolute CV risk for high-risk individuals with a baseline LDL-C routinely above 3 to 4 mmol/L. 145 Therefore, although the FOURIER and ODYSSEY OUTCOMES results have been seized upon by some as an example of expensive agents with a disappointing effect on CV endpoints, it is important for clinicians (and healthcare commissioners) to appreciate that there are high-risk patients who stand to benefit very significantly from the addition of a PCSK9i to their lipid-lowering regimen. These include individuals who have poor LDL-C control despite maximal non-PCSK9i therapy, those who are unable to tolerate high-intensity statins and those where interacting drugs limit statin dosing.
Remaining questions
Since its discovery in 2003, PCSK9 has significantly contributed to our understanding of the LDL-receptor lifecycle and the regulation of plasma LDL. In particular, we now have a more complete understanding of the genetic basis for FH. There do, however, remain a number of important unanswered questions. What benefit does PCSK9 provide given that LOF mutations do not appear to exert damaging effects, and indeed result in lower CV event rates? What role does PCSK9 play in the small intestine, kidney and pancreas? And what is the significance of its transient expression in the embryonic brain?
In the continuing efforts to combat CV disease, PCSK9 has proven a very successful drug target. The apparent lack of deleterious effects in individuals with LOF PCSK9 mutations suggests that long-term PCSK9 inhibition should be safe. And indeed the success, so far, of two monoclonal antibody inhibitors of PCSK9 (Alirocumab, Evolocumab) with regard to both LDL-C lowering efficacy, safety and tolerability is consistent with this suggestion. They have been shown to provide very significant LDL-C reductions on top of existing maximal LDL-C lowering therapy involving statin and ezetimibe combinations. This is good news for those patients who are unable to achieve LDL-C targets on oral-based regimens alone, something of particular relevance to individuals with FH. It may also mean less frequent LDL apheresis, or its cessation, in certain patients.
However, these new agents remain expensive given the life-long nature of therapy. Hence, one of the main challenges in this early PCSK9i era will be to determine which individuals may benefit most, and where PCSK9i treatment is likely to add little to existing statin therapy. For clinicians, this represents both a challenge with respect to clinical assessment but also in terms of communication to healthcare commissioners and patients.
While the principal focus of new LDL-C lowering treatment is on the mAbs, PCSK9 has proved such a fruitful target for drug development that it is relevant to speculate whether it may not be too long before these therapies are usurped by treatments requiring ever less frequent dosing. This may come to fruition relatively soon in the form of siRNA (Inclisiran) and in the more distant future, via vaccination or even gene-editing technologies. What is almost certain is that the PCSK9 story is far from over.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
JM.
Contributorship
All authors were involved in researching the literature. AP wrote the first draft of the article. All authors reviewed and edited the article and approved its final version.