
Abstract
Background
Biomarker discovery studies often claim ‘promising’ findings, motivating further studies and marketing as medical tests. Unfortunately, the patient benefits promised are often inadequately explained to guide further evaluation, and few biomarkers have translated to improved patient care. We present a practical guide for setting minimum clinical performance specifications to strengthen clinical performance study design and interpretation.
Methods
We developed a step-by-step approach using test evaluation and decision-analytic frameworks and present with illustrative examples.
Results
We define clinical performance specifications as a set of criteria that quantify the clinical performance a new test must attain to allow better health outcomes than current practice. We classify the proposed patient benefits of a new test into three broad groups and describe how to set minimum clinical performance at the level where the potential harm of false-positive and false-negative results does not outweigh the benefits. (1) For add-on tests proposed to improve disease outcomes by improving detection, define an acceptable trade-off for false-positive versus true-positive results; (2) for triage tests proposed to reduce unnecessary tests and treatment by ruling out disease, define an acceptable risk of false-negatives as a safety threshold; (3) for replacement tests proposed to provide other benefits, or reduce costs, without compromising accuracy, use existing tests to benchmark minimum accuracy levels.
Conclusions
Researchers can follow these guidelines to focus their study objectives and to define statistical hypotheses and sample size requirements. This way, clinical performance studies will allow conclusions about whether test performance is sufficient for intended use.
Introduction
Discovery of a ‘promising’ biomarker is a common rhetoric in biomarker research with claims of clinically important findings, 1 but few discoveries progress to appropriate clinical evaluation. 2 Unfortunately, what is promised in terms of clinical performance and patient benefits is often inadequately explained to guide further evaluation. An audit of diagnostic test accuracy studies has documented that few state an explicit hypothesis about the accuracy level that would favour clinical uptake, and researchers frequently over-interpret the results. 3 Such unwarranted optimism can motivate subsequent futile studies, wasting research resources. Moreover, it can lead to marketing of biomarkers as medical tests with no clinical benefits for patients or the potential for harm. 4
To date, discussion of failed clinical translation of biomarkers has primarily focused on poor study design and inadequate validation of findings.5,6 Guidelines are available to help address these problems.7–9 However, these guidelines do not include advice about determining whether biomarker performance is promising enough to justify further evaluation as a medical test. For some gaps, modest test accuracy will be sufficient to deliver clinical benefits for patients; for others, near-perfect accuracy will be required. Thus, for each potential test application, a critical question is: what level of performance is sufficient? Biomarkers performing above this level can be prioritized for further research. Poor performers can be considered futile and discarded early.
A ‘go/no-go’ threshold approach is routinely used for drug development, where a new drug only progresses to a phase 3 randomized controlled trial if it meets prespecified minimum levels of safety and efficacy in a phase 2 study. 10 It has also been proposed for biomarker development by using a decision-analytic framework to set minimum accuracy levels.11,12 However, the concepts are not yet well understood by non-statisticians and it is not commonly used, nor required by regulatory agencies for premarket approval of new tests. So far, regulatory agencies, such as the Food and Drug Administration in the United States and the Conformité Européene marking body in the European Union (EU) require that new biomarkers meet analytical performance specifications, and it has been possible to market tests based on safety with limited clinical evidence. However, this situation is changing with recent in vitro diagnostic (IVD) devices directives requiring more evidence on clinical performance for intended use. 13
To address this problem, members of the European Federation of Clinical Chemistry and Laboratory Medicine’s Test Evaluation Working Group have developed a step-by-step guide on the practical aspects of setting clinical performance specifications from a clinical decision-making perspective.
Methods
Definition and approach
We define clinical performance specifications as a set of criteria that quantify the clinical performance a new test must attain to allow better health outcomes than current practice.
We used established test evaluation and decision-analytic frameworks to develop the guide and piloted it on examples at three face-to-face meetings. Explanations of the concepts and approach were refined at two training workshops attended by clinicians, laboratory scientists, IVD industry, health economists, health technology assessment and government re-imbursement agencies.
Test evaluation framework
To justify implementation as a medical test, a biomarker must lead to improved health outcomes, or provide other benefits over existing tests without compromising health outcomes, such as greater patient convenience, simplifying the health-care process or reducing resource use. 7
After discovery of an association between the biomarker and a clinical condition or state, sometimes referred to as evidence of ‘scientific validity’, 14 the five components for evaluation as a medical test are: analytical performance – the technical ability of the test assay/device to measure the biomarker, sometimes referred to as ‘analytical validity’; clinical performance – the ability to provide information about the condition or state of interest for its intended use in the relevant population, sometimes referred to as ‘clinical validity’; clinical effectiveness – the ability to improve health outcomes over existing tests; cost-effectiveness and broader impact of use including societal consequences. 7
The initial focus for an evaluation is analytical and clinical performance as required for regulatory approval. Both have an impact on clinical effectiveness, but good analytical and clinical performance do not of themselves mean that the test will lead to improved patient outcomes. By prespecifying the minimum clinical performance levels needed to achieve the proposed clinical benefits, researchers can design more purposeful clinical performance studies to determine if test performance is sufficient for intended use.
Measures of clinical performance include test accuracy which is traditionally expressed as sensitivity and specificity and by estimating the positive predictive value (PPV) of a positive test result and negative predictive value (NPV) of a negative test result (Supplementary Figure). PPV and NPV vary across populations with different disease prevalence. Sensitivity and specificity can also vary between populations due to differences in patient spectrum.15,16 Thus, to provide meaningful estimates of clinical performance, studies of test accuracy need to be conducted in a population and in a well-defined clinical pathway that closely reflects how the test is intended to be used in practice.
Decision-analytic framework
Taking a clinical decision-making perspective, the minimum acceptable clinical performance for a test represents the accuracy level above which the intended clinical benefits outweigh the potential harms. 12 Fundamental to this approach is that patients have to benefit from testing, and that testing primarily affects patient-important health outcomes through the way test results and findings are used to guide downstream clinical actions.
To draw the link between test accuracy and health outcomes, one must define the clinical consequences of true/false positive and negative results for the target condition, relative to current practice without the new test. These are the clinical decisions (actions) triggered by the test result and potential consequences for patient health outcomes. In broad terms, a new test that detects more true positives (TP) than existing tests may improve disease outcomes by allowing improved treatment for individuals with a positive result; but may also detect more false positives (FP) leading to iatrogenic harm through unnecessary further testing or treatment. We define an FP as a finding that is subsequently confirmed to be incorrect; to distinguish it from over-diagnosis, a pathologically ‘correct’ TP that does not represent clinically significant disease.
Conversely, a test that detects more true negatives (TN) may reduce iatrogenic harm by allowing avoidance of further tests or unnecessary treatment for individuals with a negative result, but may also detect more false negatives (FN), leading to worse disease outcomes through the missed opportunity for treatment.
Our guide is based on the decision-analytic principles described by Vickers et al. who have expressed the trade-off between benefit and harm of a test as the ‘net benefit’ (benefit–harm).17,18 In its simplest form, the net benefit can be calculated as (TP/N–FP/N), where N=number tested. It relies on the assumption that a TP leads to health benefits and requires the health consequences of TP and FP to be judged on the same metric. This can be done by defining the number of FP that would be tolerated to identify one TP (‘FP:TP threshold’) and weighting the FP/N proportion accordingly, so the unit of net benefit is one TP. 18 By definition, a test that exceeds the minimum acceptable clinical performance levels achieves a net benefit >0. The net benefit does not provide an estimate of the actual health consequences of a TP finding, e.g. longer survival time.
Results
Practical guide
We describe a five-step approach for setting clinical performance specifications. The approach and examples are summarized in Figure 1 and Table 1.
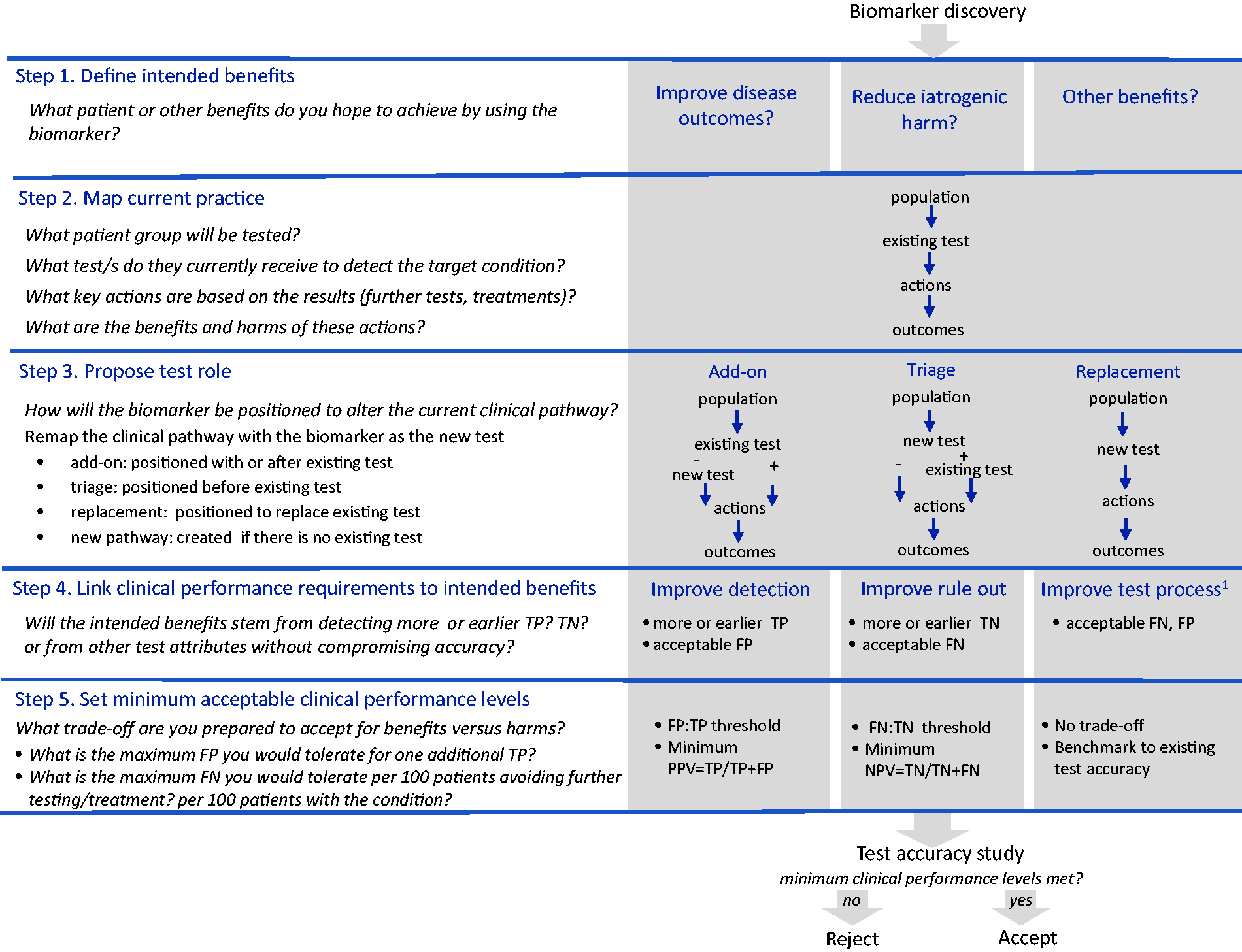
Step-by-step guide for setting clinical performance specifications.
Illustrative examples for setting clinical performance specifications.
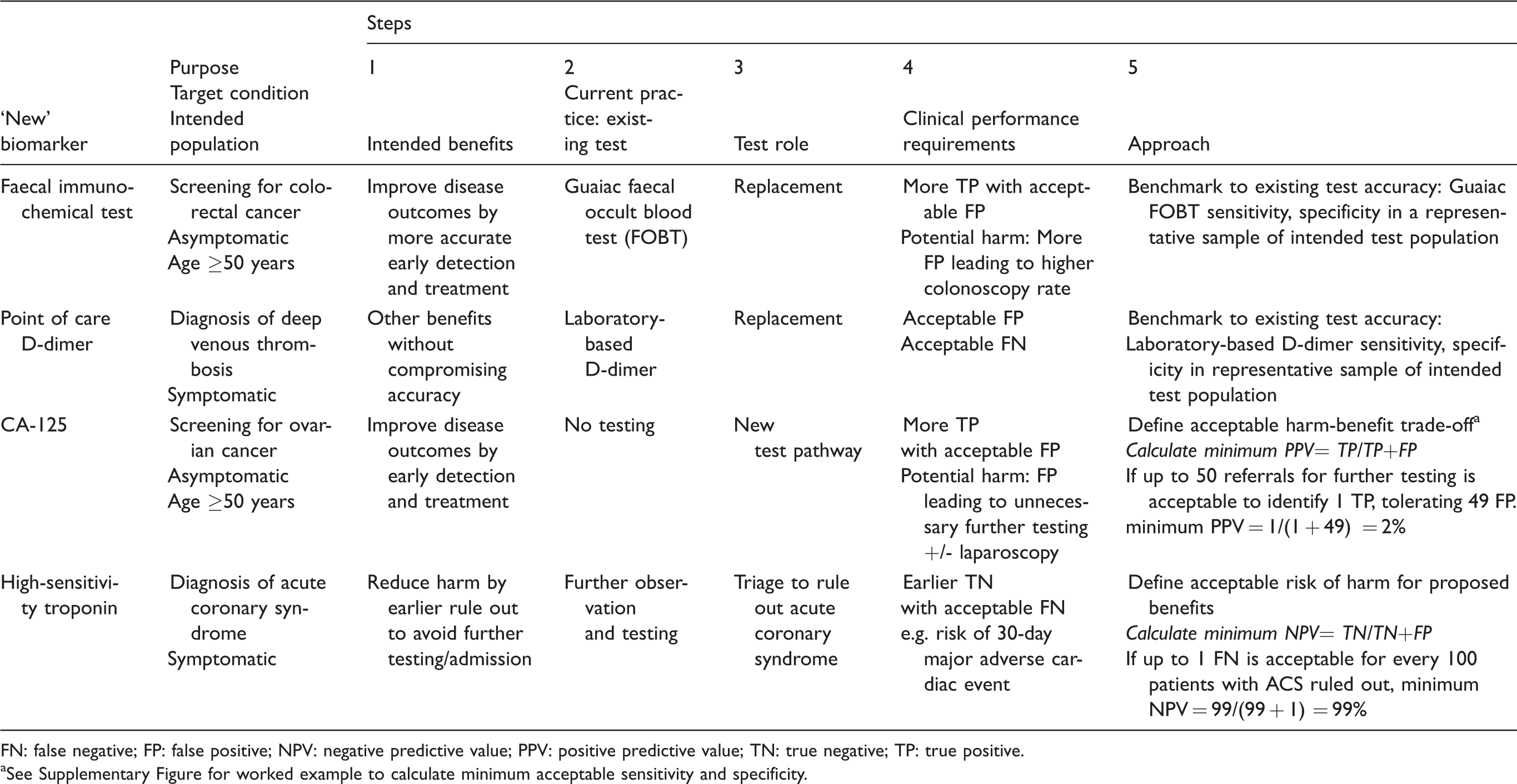
FN: false negative; FP: false positive; NPV: negative predictive value; PPV: positive predictive value; TN: true negative; TP: true positive.
aSee Supplementary Figure for worked example to calculate minimum acceptable sensitivity and specificity.
Step 1. Define the intended benefits
Researchers can start the process by bringing the findings of a biomarker discovery study to relevant clinical groups to identify the target condition, the population for testing and the intended benefits for patients or others. Ask ‘What patient or other benefits do you hope to achieve by using the biomarker?’ We classify the potential benefits of a new test into three broad categories that can be used as prompts: (1) ‘Will the test improve disease outcomes?’, e.g. by allowing more accurate or earlier detection of the target condition that will benefit from treatment; (2) ‘Will the test reduce iatrogenic harm?’, e.g. by offering more accurate, earlier or less invasive rule-out of the condition, so patients can avoid unnecessary further tests and treatment or (3) ‘Will the test provide other benefits?’, e.g. by improving patient or provider experience such as the convenience of the testing process, or reduced costs, without compromising accuracy.
Step 2. Map current practice
Defining the test purpose and desired changes in outcomes requires a close understanding of the current clinical pathway. We recommend drawing a simple flowchart of current practice with input from relevant clinician groups. 19 This flowchart should describe existing tests, if any; the key actions informed by the test results, such as initiation, cessation or change of treatment or use of further tests and the health outcomes of these actions.
Step 3. Propose test role
Redraw the clinical pathway to show where the new test will be positioned to achieve the intended benefits. Possible roles are as an add-on, triage or replacement test 20 ; or if no existing test, as a new test pathway. If intended to improve disease detection, ask ‘Will the new test replace or be an add-on to the existing test(s)?’ If intended to reduce the use of other tests or treatment, ask ‘Where will it be positioned in the clinical pathway to allow triage of patients to avoid further testing and management?’ If intended to provide other benefits, ask ‘Is it intended to replace the existing test without comprising accuracy?’
Step 4. Link clinical performance requirements to intended benefits
One can work back from the desired changes in outcomes to identify the clinical performance requirements by asking ‘Will the intended benefits stem from detecting more or earlier TP than the existing test? Or TN? Or from other test attributes without compromising accuracy?’ For example, if the biomarker is intended to improve disease outcomes by improving diagnosis and treatment, the intended benefits stem from actions following a positive test result. Here, the new test strategy must demonstrate more or earlier TP findings than the existing test strategy with an acceptable number of FP. If intended to reduce harm by avoiding further testing and treatment, the benefits stem from a negative test result. Here, the new test must demonstrate more TN, or be positioned before the existing test strategy to allow earlier rule out than existing tests with an acceptable number of FN. If proposed as a replacement test with other benefits, the new test must demonstrate these benefits with an acceptable number of FN and FP.
Step 5. Set minimum acceptable clinical performance levels
To set minimum acceptable clinical performance levels, ask ‘What harm–benefit trade-off are you prepared to accept?’ Here, it is desirable to seek input from clinicians, patients, policy-makers and other stakeholders.
Examples
Improve disease outcomes – FP:TP trade-off
For biomarkers intended to improve disease outcomes, the trade-off is between the proposed benefit of TP and potential harm of undergoing unnecessary further tests or treatment due to FP. To elicit the minimum acceptable trade-off, start by describing the clinical consequences of a TP and an FP, then ask ‘What is the highest number of individuals having a FP that you would be prepared to accept for one additional individual to have the benefit of a TP finding?’ Apply this FP:TP threshold to set the minimum acceptable PPV (PPV=TP/TP+FP). A test that does not meet this threshold in test accuracy studies can be rejected from further evaluation.
An example is biomarkers for screening for ovarian cancer, where the intended benefit of a TP is improved survival by detecting asymptomatic cancer at an early stage when treatment is potentially more effective. The potential harm of an FP includes unnecessary anxiety and further testing such as intravaginal ultrasound, potentially leading to laparotomy to rule out cancer with the risk of surgical complications. Using this information, if it is considered acceptable to detect one cancer for every 50 women testing positive, i.e. 49 women receive a false alarm of potential ovarian cancer and require further testing for each case of cancer detected early (FP:TP = 49:1), then the minimum acceptable PPV is 2%. Candidate biomarkers such as CA-125 with a PPV below this value can be rejected from further evaluation as a population screening test. There is no need to set a minimum NPV if it is reasonable to assume the consequences of a FN are not more harmful than no screening.
Given current practice is no screening (TP = 0), a biomarker with low sensitivity will still warrant further evaluation if it meets the prespecified FP:TP threshold. Pepe et al. present worked examples to show how to calculate minimum acceptable sensitivity and specificity combinations that meet the FP:TP threshold. 12 This calculation requires an estimate of the disease prevalence (Supplementary Figure).
The FP:TP threshold will vary in different settings according to the consequences of an FP. For ovarian cancer screening, others have set this threshold at 9:1 if a positive test triggers a laparotomy, to arrive at a minimum PPV of 10%.21,22
For tests achieving minimum clinical performance levels, further evaluation is required to confirm the claims of improved disease outcomes for TP. While threshold setting presupposes there is some evidence that patients with the condition will benefit from treatment or other actions such as counselling; this evidence is usually for patients diagnosed with the condition using standard tests. It is pertinent to ask if the additional TP identified by the new test will receive a treatment advantage for (early) detection versus (delayed) diagnosis without the new test. For ovarian screening tests, definitive evidence from clinical trials is still needed to confirm the proposed treatment advantage from earlier detection. If the additional TP represent a broader spectrum/definition of disease and would otherwise remain undetected without the new test, the potential for over-diagnosis exists. 23 Here, pertinent questions for further evaluation are whether the additional cases are clinically important or whether the harms of medical labelling and intervention outweigh any benefits. In these situations, clinical trials will be needed to provide definitive evidence that the benefits of testing and subsequent actions outweigh the harms.
Reduce iatrogenic harm – FN:TN trade-off
For biomarkers intended to rule out disease, the test negativity rate indicates the maximum proportion of patients who could benefit from rule out to avoid unnecessary further testing or treatment. The main potential for harm is an FN result. Start by describing the clinical consequences of a TN and FN, then ask ‘How many FN are you prepared to tolerate for every 100 (or 1000) patients ruled out?’ This value can be used to set the minimum acceptable FN:TN threshold for patient safety and the NPV (NPV=TN/TN+FN). Given all patients would receive further testing or treatment under standard practice, a new test that meets the FN:TN threshold will additionally be required to demonstrate a minimum acceptable sensitivity. This can be set by asking ‘How many FN are you prepared to tolerate for every 100 patients with the condition? One should check the frequency of FN under standard practice without the new test when setting this value.
In the example of new tests to rule out acute coronary syndrome (ACS) in patients presenting to the emergency department with chest pain, Than et al. reported on a clinician survey to elicit the acceptable risk of FN. 24 In this survey, the potential consequences of a FN were described as a missed major adverse cardiac event (MACE) within 30 days of discharge. Almost half the emergency department clinician respondents considered up to 1 missed MACE per 100 patients to be acceptable. 24 These findings support a minimum acceptable NPV of at least 99%, i.e. no more than 1 FN for every 100 patients eligible for discharge based on a negative test result. The authors concluded that a new triage test should also be required to demonstrate a minimum acceptable sensitivity of at least 99% – no more than 1 missed diagnosis per 100 patients with ACS.
One should also consider the potential harms of a FP. These are generally less consequential than an FN for rule-out tests; in particular, if a positive result leads to further testing, the same as would occur without the new test.
Other benefits – Benchmark to existing tests
For biomarkers intended to provide other benefits without compromising accuracy, minimum test accuracy levels can be benchmarked against the existing test. For example, faecal immunochemical testing (FIT) was proposed to replace guaiac faecal occult blood testing (FOBT) for screening to improve early detection of colorectal cancer (TP) without increasing the rate of unnecessary colonoscopies (FP). 25 Here, minimum accuracy levels could be set to require sensitivity higher than the existing guaiac test with non-inferior specificity. Alternatively, if the major proposed benefit was greater adherence to testing due to no dietary restrictions, then minimum sensitivity and specificity levels could be set at the same level as the guaiac test. A study of adherence to testing would also be needed to provide evidence of the intended benefits.
Another example is point-of-care D-dimer in primary care to replace laboratory testing in low-risk patients with suspected deep venous thrombosis, where the intended benefit is improved patient and provider convenience. 26 The clinical sensitivity and specificity of the laboratory test provide a benchmark for point-of-care tests. Here, the convenience of point-of-care testing may come at the expense of analytical performance which can impact clinical accuracy. If a trade-off will be tolerated between the proposed benefits of the test and the potential harms of more FP or FN, benchmarking is not suitable; one can follow the approaches outlined above to deal with this trade-off.
Benchmarking can also be used for new tests proposed for cost savings without compromising accuracy. If the minimum clinical performance requirements are met, then an economic analysis of the new test and all downstream costs versus current practice would be needed to demonstrate the proposed benefits. If the cost savings are proposed at the expense of clinical performance, benchmarking is not suitable. Here, minimum clinical performance levels can be set by defining an acceptable safety threshold for FN as described above for rule out tests.
The Supplementary Material outlines how the same approach can be used for tests for other purposes beyond diagnosis and screening.
Discussion
We invite researchers to use this guide for setting minimum clinical performance levels to strengthen clinical performance study design, interpretation and reporting. By following this approach, researchers are pushed to seek information from clinical groups, patients and other stakeholders about the potential consequences of test results compared to current practice without the test and judge the clinical benefits versus harms that would support translation. The major advantage is to allow an explicit and early determination of whether or not biomarker performance warrants further evaluation as a medical test, and to reduce research waste and inappropriate clinical use of poor performers.
We advocate setting the minimum clinical performance level to formulate a study hypothesis that clinical performance is adequate for intended use. This is analogous to setting the ‘minimum clinically important difference’ (the smallest change in an outcome that a patient would identify as important) for a trial of a new treatment. One should calculate the study sample size to provide adequate precision to test this hypothesis and include this information when reporting study results to meet the STAndards for the Reporting of Diagnostic accuracy studies (STARD) guideline requirements. 8 A finding of test accuracy above the minimum acceptable level supports the conclusion that clinical performance is sufficient for intended use. Researchers can also use this guide when developing performance evaluation plans to meet new EU regulations for IVDs. 13
The decision-analytic principles for setting clinical performance specifications for tests are well established.11,12 In this paper, we focus on the practical aspects with the aim to promote wider awareness and uptake of these methods among laboratory, clinical and industry researchers. We provide a step-wise approach with trigger questions to help guide collaboration between these groups. The importance of early clinician input is shown by the example of high-sensitivity troponin as a triage test for ACS assessment, which may have been rejected if safety concerns focused on the frequency of FP rather than FN due to a poorly defined clinical pathway.
Where the clinical performance specifications are not met, laboratory professionals can advise on the potential for further development to improve analytical performance to meet these clinical needs. For illustration, we have presented examples of single biomarker tests; however, the principles also apply to biomarker panels and multiplex ‘omics’ as described by Skates et al., 27 where the need for efficient screening of candidate biomarkers is very high.
For biomarkers with multiple potential indications, setting clinical performance specifications for each indication may help prioritization. For example, initial studies demonstrating an association between procalcitonin and bacterial infection 28 motivated wide clinical interest in its use to guide antibiotic decisions for a range of indications. Regulatory approval followed, however, in the critical care setting, translation stalled despite accumulating evidence of test accuracy, with debate about appropriate indications for use for patients with suspected or confirmed sepsis.29,30 Setting a priori performance specifications for each potential indication (e.g. withhold antibiotic therapy, stop antibiotic therapy early) to guide study design and interpretation could help expedite definitive evaluation.
The trade-off between benefit and harm is a value judgement and can be expected to vary among clinicians, patients and policy-makers. It may also vary between health-care settings due to considerations, such as cost and clinical service capacity. Researchers should therefore explain who participated in setting the minimum level and how it was arrived at. The goal is not to reach a single point of agreement, but rather to ensure the level set is transparent. If the value varies substantially between groups, it can be set at a level low enough that most would agree a test not meeting this level is not worth pursuing. While this is a basic approach to standard setting, we argue it is less flawed than the typical approach of interpreting accuracy estimates with no a priori defined levels. As an extension, researchers can use the methods described by Vickers et al. to plot a ‘decision curve’ of the net benefit of current practice strategies (existing tests, no tests) across a plausible range of acceptable FP:TP thresholds. 18 The clinical performance specifications for a new biomarker to improve on current practice can then be read from the plot at the threshold values of interest to different individuals and groups.
Some trade-offs will be more challenging to deal with than those discussed here. For example, where there are multiple important benefits and harms to weigh up or the potential harms will be borne by patients but not the proposed benefits, e.g. cost savings to the health system. In these situations, more complex approaches may be needed, such as discrete choice experiments and multicriteria decision-analytic models.
Another challenge is that for some indications, it will not be possible to reach agreement among clinicians about the current clinical pathway. The clinical pathway will also vary between countries with different health-care resources. A pragmatic approach is to select the health-care setting; patient group, existing tests and actions most likely to favour the new test and set the minimum performance levels for this ‘best case scenario’.
A potential criticism for adopting this approach in the early stages of biomarker development is that directing evaluation toward a predefined clinical indication will stifle innovation and preclude the discovery of broader biological insights. While studies assessing the association between a biomarker and normal or pathological processes are essential to advance knowledge of disease biology and identify potential treatment targets, we argue that these need to be distinguished from studies designed to develop and evaluate medical tests. Conflating these study purposes is counter-productive and an important source of research waste, as demonstrated by the extensive evaluation of cancer biomarkers, such as p53 31 published in clinical journals, which are yet to find a role in routine clinical practice.
For tests meeting minimum clinical performance levels, validation in an independent sample is still required. In some cases, this evidence will be sufficient for conclusions about improved health outcomes. An example is FIT as a replacement for guaiac-based FOBT, where the clinical benefits of increased TP and TN are well established. Where uncertainty exists, achieving minimum performance levels will be necessary but not sufficient and randomized trials may be needed. 32 In particular, for tests proposed to improve disease outcomes, a randomized controlled trial will be needed to demonstrate the effectiveness of management in patients with the biomarker-defined condition who would have otherwise gone undetected using the current test strategy. For definitive evidence about both benefits and harms, trials comparing the new test strategy versus standard care with follow-up to assess health outcomes are required, as have been performed for test strategies for ovarian cancer screening 33 and rule-out of ACS. 34
Finally, the approach described demands early collaboration and crosstalk between biomarker discovery researchers, clinicians, laboratory professionals, the IVD industry, as well as patients and health policy makers. We hope this paper can help facilitate these interprofessional discussions and lead to more efficient biomarker translation to clinical practice.
Supplemental Material
Supplemental Material1 - Supplemental material for Setting clinical performance specifications to develop and evaluate biomarkers for clinical use
Supplemental material, Supplemental Material1 for Setting clinical performance specifications to develop and evaluate biomarkers for clinical use by Sarah J Lord, Andrew St John, Patrick MM Bossuyt, Sverre Sandberg, Phillip J Monaghan, Maurice O’Kane, Christa M Cobbaert, Ralf Röddiger, Lieselotte Lennartz, Cecilia Gelfi, Andrea R Horvath and for the Test Evaluation Working Group of the European Federation of Clinical Chemistry and Laboratory Medicine in Annals of Clinical Biochemistry
Supplemental Material
Supplemental Material2 - Supplemental material for Setting clinical performance specifications to develop and evaluate biomarkers for clinical use
Supplemental material, Supplemental Material2 for Setting clinical performance specifications to develop and evaluate biomarkers for clinical use by Sarah J Lord, Andrew St John, Patrick MM Bossuyt, Sverre Sandberg, Phillip J Monaghan, Maurice O’Kane, Christa M Cobbaert, Ralf Röddiger, Lieselotte Lennartz, Cecilia Gelfi, Andrea R Horvath and for the Test Evaluation Working Group of the European Federation of Clinical Chemistry and Laboratory Medicine in Annals of Clinical Biochemistry
Footnotes
Acknowledgements
We are grateful to Wilma DJ Verhagen-Kamerbeek, Sarah Robinson, Ann Incamps, Michael Hausmann and Tomas Salek from the EFLM Test Evaluation Working Group for their valuable comments on earlier drafts of this article. Neither the funding organization (EFLM) nor the independent educational support by Roche Diagnostics, Abbott and Thermo Fisher Scientific influenced the content of this paper. This publication reflects the collective view of the Working Group. The authors include representatives from Roche Diagnostics and Abbott Diagnostics as they provided important insights from the IVD industry, a key stakeholder in the research and development of new biomarker assays.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors declare support for travel to meetings from The European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) for the submitted work. One author (RR) declares a financial relationship with Roche Diagnostics (RR) as a salaried employee; and one author (LL) declares a financial relationship with Abbott as a salaried employee. As in vitro diagnostic (IVD) industry stakeholders, these organizations might have an interest in the submitted work. Roche Diagnostics, Abbott and Thermo Fisher Scientific have provided independent educational grants to EFLM.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) provided financial support to the Test Evaluation Working Group for the preparation of this paper through payment of travel expenses to attend Working Group meetings. Abbott, Roche Diagnostics, and Thermo Fisher Scientific provide independent educational grant to EFLM (EFLM grant reference number 2017/01, 2017/02 and 2017/04, respectively).
Ethical approval
Not applicable.
Guarantor
SJL.
Contributorship
All authors contributed to the paper as members of the European Federation of Clinical Chemistry and Laboratory Medicine Test Evaluation Working Group (EFLM-TEWG). ARH and SS initiated the topic for development of methodological guidelines by the EFLM-TEWG. SJL led the discussions for the development of the paper. SJL, AS-J, PMMB, SS, PJM, MO’K, CMC, RR, LL, CG and ARH contributed to the development of the ideas and examples. PMMB helped to define the concepts and refine the scope. SJL and AS-J prepared the first draft. All authors contributed to article revisions.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.