
Abstract
Background
Hypomagnesaemia is present in 40–50% of children with autosomal dominant renal cysts and diabetes syndrome (RCAD). On the contrary, the prevalence of hypomagnesaemia in children with autosomal dominant polycystic kidney disease (ADPKD) has never been examined. We aimed to investigate whether hypomagnesaemia is present in children with polycystic kidney diseases.
Methods
Children with cystic kidney diseases were investigated in a cross-sectional study. Serum concentrations of magnesium (S-Mg) and fractional excretion of magnesium (FE-Mg) were tested. Fifty-four children with ADPKD (n = 26), autosomal recessive polycystic kidney disease (ARPKD) (n = 16) and RCAD (n = 12) with median age of 11.2 (0.6–18.6) years were investigated.
Results
Hypomagnesaemia (S-Mg < 0.7 mmol/L) was detected in none of the children with ADPKD/ARPKD and in eight children (67%) with RCAD. Median S-Mg in children with ADPKD/ARPKD was significantly higher than in children with RCAD (0.89 vs. 0.65 mmol/L, P < 0.01). The FE-Mg was increased in 23% of patients with ADPKD/ARPKD (all had chronic kidney disease stages 2–4) and in 63% of patients with RCAD, where it significantly correlated with estimated glomerular filtration rate (r = −0.87, P < 0.01).
Conclusions
Hypomagnesaemia is absent in children with ADPKD or ARPKD and could serve as a marker for differential diagnostics between ADPKD, ARPKD and RCAD in children with cystic kidney diseases of unknown origin where molecular genetic testing is lacking. However, while hypomagnesaemia, in the absence of diuretics, appears to rule out ADPKD and ARPKD, normomagnesaemia does not rule out RCAD at least in those aged <3 years.
Keywords
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) caused by mutations of PKD1 (polycystic kidney disease 1) or PKD2 (polycystic kidney disease 2) genes is the most common monogenic form of kidney disease with a prevalence of 1:500–1000. 1 Renal cysts and diabetes syndrome (RCAD) caused by deletion or mutation of the HNF1B (hepatocyte nuclear factor 1 homeobox B) gene is also an autosomal dominant cystic kidney disease. However, in about 50% of the cases, de novo deletions/mutations are present in the patients causing negative family history of kidney disease. Therefore, negative family history of cystic kidney disease in parents does not rule out RCAD. Furthermore, a negative parental history of renal cysts is a diagnostic hallmark of another cystic kidney disease in childhood, namely autosomal recessive polycystic kidney disease (ARPKD) caused by mutations of a single gene PKHD1 (polycystic kidney and hepatic disease 1). 2 Therefore, differential diagnosis between ADPKD, ARPKD and RCAD could sometimes be difficult in children with cystic kidney diseases where molecular genetic tests for anomalies in HNF1B, PKD1, PKD2 and PKHD1 genes are lacking.
Hypomagnesaemia is present in 40–50% of children with autosomal dominant renal cysts and diabetes syndrome (RCAD). 3 On the contrary, the prevalence of hypomagnesaemia in children with ADPKD has never been examined and has been found in only 5% of children with ARPKD in one previous study. 4 The aim of the study was to investigate whether hypomagnesaemia is present in children with ADPKD or ARPKD.
Materials and methods
Patients
All 26 children with ADPKD, 16 children with ARPKD and 12 children with RCAD followed up in our two tertiary paediatric nephrology centres during the years 2013–2014 were investigated. One child with ADPKD was excluded from the study because of the use of a thiazide diuretic (for hypertension together with an ACE-inhibitor) that is known to cause hypomagnesaemia. 5
The demographic parameters of the patients are summarized in Table 1.
Demographic data of children with ADPKD, ARPKD and RCAD.
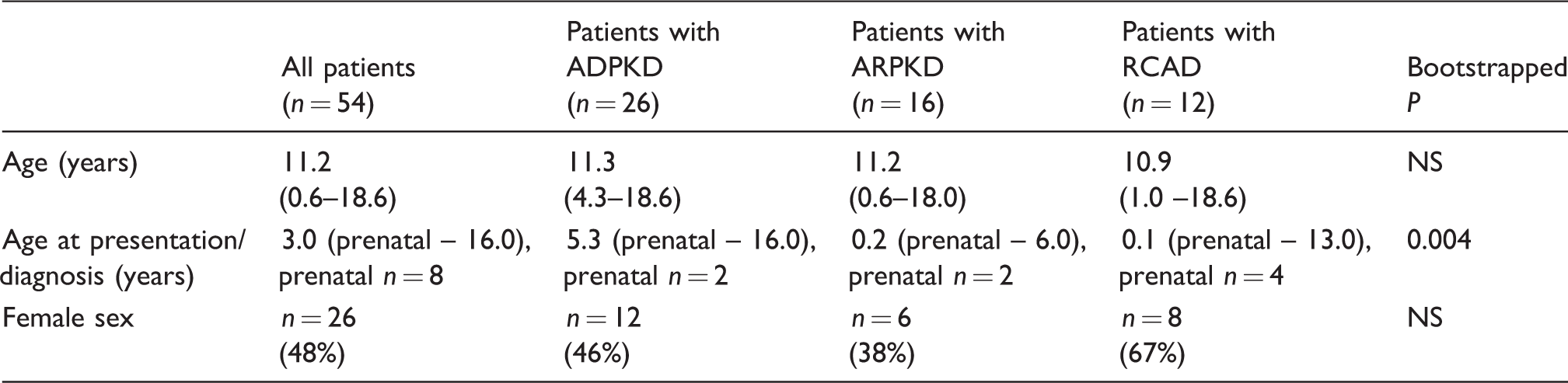
Note: Data are given as median (range) or as relative frequency (%).
ADPKD: autosomal dominant polycystic kidney disease; ARPKD: autosomal recessive polycystic kidney disease; RCAD: renal cysts and diabetes syndrome.
The diagnosis of ADPKD was based on positive family history of ADPKD (affected father in 15 cases) in all children combined with at least three renal cysts on ultrasound. 6 The diagnosis of ADPKD was supported by positive result from molecular genetic analysis in six children (positive linkage analysis for PKD1 in two children, identification of PKD1 or PKD2 gene mutations in three and one patient, respectively), in the remaining 20 children molecular genetic analysis was not performed.
The diagnosis of ARPKD was based on established clinical criteria including: (1) typical kidney involvement on ultrasound (enlarged hyperechogenic kidneys with bilateral poor corticomedullary differentiation); (2) typical liver involvement (congenital hepatic fibrosis, ductal plate malformation) and (3) normal renal US of both parents, consistent with autosomal-recessive inheritance. 2 The diagnosis of ARPKD was supported by positive result from molecular genetic analysis (identification of PKHD1 gene mutations) in nine children, in the remaining seven children, molecular genetic analysis was not performed.
The diagnosis of RCAD was based solely on positive result from molecular genetic analysis of the HNF1B gene in all patients (whole gene deletion in eight children, mutation in four children). De novo deletion/mutations were present in eight children. The renal phenotype of children with RCAD was very heterogeneous ranging from bilateral polycystic kidney involvement to multiple cysts in a functional solitary kidney. Two children have maturity-onset diabetes of the young and were treated with insulin. Three children with RCAD were already treated with magnesium supplementation for a known hypomagnesaemia. No child was treated with thiazide or loop diuretics (exclusion criteria).
All patients and/or their parents signed informed consent forms. All samples were analysed blindly, and the procedures were in accordance with the Declaration of Helsinki. This study was approved by the Ethical Committee of The Internal Grant Agency of the Czech Ministry of Health (IGA-MZ ČR).
Laboratory investigations
The serum and urinary concentrations of total magnesium were determined with a colorimetric method with xylidine blue; the serum and urinary concentrations of total calcium were determined with a colorimetric method with Arsenazo-III (automatic analyser Advia 1800, Siemens). The serum calcium concentrations were not albumin adjusted. Hypomagnesaemia was defined as total magnesium concentration < 0.7 mmol/L or treatment with magnesium supplements due to hypomagnesaemia in the past, and, hypermagnesaemia, as total magnesium concentration > 1.0 mmol/L. Standard formulas were used for the determination of the fractional excretion of magnesium and calcium (FE-Mg, FE-Ca). Increased FE-Mg was defined as FE-Mg > 4% in patients with hypomagnesaemia and > 5% in patients with normal serum magnesium concentrations. 4 Hypocalciuria was defined as FE-Ca < 1%. Serum creatinine concentrations were determined by the enzymatic method (automatic analyser Advia 1800, Siemens). Glomerular filtration rate was estimated (eGFR) using the Schwartz formula. 7 The stage of chronic kidney disease was assessed using the definition of the National Kidney Foundation’s Disease Outcomes Quality Initiative Clinical Practice Guidelines. 8
Statistical analysis
The results are given as frequencies or median (range) for non-normally distributed data. Statistical analysis was performed using R statistical package. 9 In order to overcome the small size of the groups, the bootstrapped ANOVA method with 1000 repeats has been deployed for testing the between group difference, using the boot package. Bootstrapped correlation coefficients were used to assess the correlation between characteristics.10,11 P < 0.05 was regarded as a statistically significant difference. The cut-off value has been determined using the standard procedure recommended for clinical biochemistry.12,13
Results
Hypomagnesaemia was detected in none of the children with ADPKD/ARPKD and in eight children (67%) with RCAD including three with Mg supplements. The median S-Mg in children with ADPKD/ARPKD was significantly higher than in children with RCAD. The data are given in Table 2. Using these data, we have determined the S-Mg cut-off value for RCAD to be S-Mg < 0.66 mmol/L with accuracy of 0.943. In case of setting the cut-off value to the decision limit of S-Mg <0.7 mmol/L, the accuracy decreased only to 0.932.
Laboratory findings in children with ADPKD, ARPKD and RCAD.
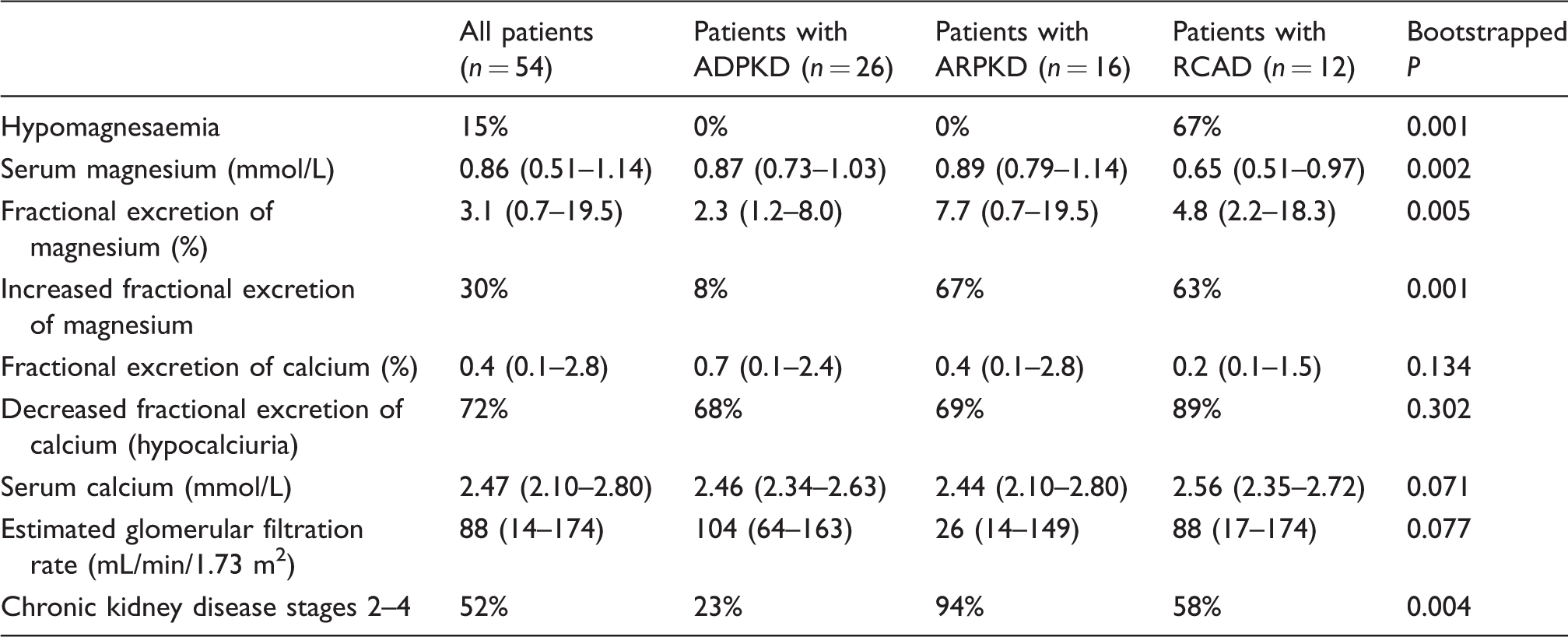
Note: Data are given as median (range) or as relative frequency (%). Hypomagnesaemia (serum magnesium < 0.7 mmol/L), increased fractional excretion of magnesium (FE-Mg > 4% in hypomagnesaemic and > 5% in normomagnesaemic patients), hypocalciuria (FE-Ca < 1%).
ADPKD: autosomal dominant polycystic kidney disease; ARPKD: autosomal recessive polycystic kidney disease; RCAD: renal cysts and diabetes syndrome.
Five children (four with ARPKD – all with decreased eGFR and one with ADPKD with normal eGFR) had hypermagnesaemia.
The FE-Mg was increased in 23% of patients with ADPKD/ARPKD (all had chronic kidney disease stages 2–4) and in 63% of patients with RCAD.
A negative correlation between FE-Mg and eGFR and FE-Ca and eGFR was found in children with RCAD but not in children with ADPKD/ARPKD. A positive correlation between FE-Mg and FE-Ca was found in children with RCAD and ARPKD. There was no correlation between serum magnesium and other assessed parameters and characteristics (e.g. FE-Mg, FE-Ca, eGFR) in either of patients’ groups. The correlation coefficients are given in Table 3.
Correlation coefficients in children with ADPKD, ARPKD and RCAD.
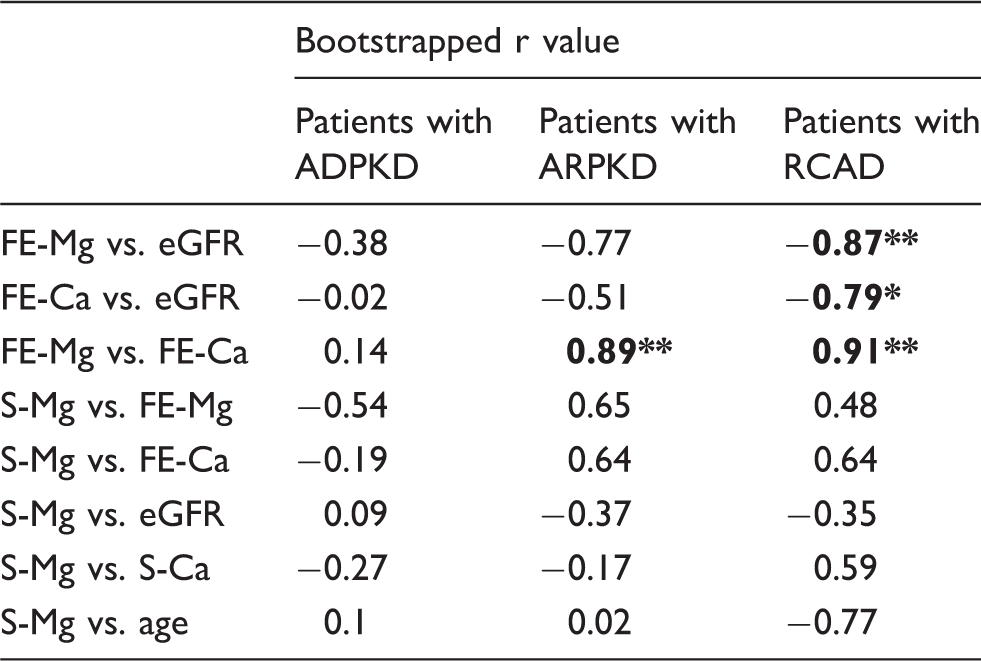
Note: Statistically significant correlation coefficients are highlighted in bold, **P < 0.01, *P < 0.05.
ADPKD: autosomal dominant polycystic kidney disease; ARPKD: autosomal recessive polycystic kidney disease; RCAD: renal cysts and diabetes syndrome; FE-Mg: fractional excretion of magnesium; FE-Ca: fractional excretion of calcium; eGFR: estimated glomerular filtration rate; S-Mg: serum magnesium; S-Ca: serum calcium.
All screened children with RCAD below the age of three years had normal magnesaemia on contrary to no child older than three years.
eGFR, serum calcium, FE-Ca, and, the proportion of children with chronic kidney disease stages 2–4 and hypocalciuria, are given in Table 2.
Discussion
Several studies have shown that hypomagnesaemia is present in 40–50% of patients with RCAD.3,14 It has become an important screening test for evaluation of the risk of having RCAD. 15
On contrary to RCAD, there is very limited information on magnesium concentrations in patients with ARPKD and ADPKD. In the only paediatric study referring serum magnesium concentrations so far, hypomagnesaemia has been found in only 5% of children with ARPKD. 4
One study in adult patients with ADPKD has found significantly lower concentrations of magnesaemia. 16 However, the authors did not give information about the prevalence of hypomagnesaemia. Furthermore, they included also patients with diuretics – a drug that is known to cause hypomagnesaemia; 5 therefore, lower concentrations of magnesaemia due to diuretic therapy cannot be excluded.
In children with ADPKD, serum magnesium has never been examined; therefore, our study is the first study investigating magnesaemia in children with ADPKD and only the second testing magnesaemia in children with ARPKD. We have found that, on contrary to children with RCAD, that all children with ADPKD and ARPKD have normal (or elevated) serum concentrations of magnesium. Therefore, in our cohort, hypomagnesaemia ruled out the presence of ADPKD or ARPKD provided the patients were not on diuretics and can be used as a sensitive marker with very high negative predictive value in the differential diagnostic of ADPKD, ARPKD and RCAD in children with cystic kidney disease of unknown origin. The only child with ADPKD and hypomagnesaemia was excluded from the study because of the use of diuretic therapy that is known to cause hypomagnesaemia. 5
Significant negative correlation between fractional excretion of magnesium and eGFR was found in children with RCAD. The fractional excretion of magnesium was found to be normal in all children with ADPKD/ARPKD who had normal eGFR, i.e. with chronic kidney disease stage 1. On the other hand it was often increased together with hypermagnesaemia in children with decreased eGFR. This finding is consistent with magnesium retention together with increased fractional excretion in the remnant functioning nephrons when eGFR is decreased below 25% of normal. 17 Therefore, decreased eGFR together with impaired total Mg excretion could potentially mask hypomagnesaemia in children with ARPKD who had decreased eGFR.
Hypocalciuria was present in nearly all children with RCAD and in nearly 70% of children with ADPKD/ARPKD. The fractional excretion of calcium significantly negatively correlated with eGFR in children with RCAD, and, also in this patient group, significant positive correlation between FE-Mg and FE-Ca was present. Hypocalciuria was also present in the index case with RCAD and hypomagnesaemia described by Adalat et al., 3 and in both children studied by van der Made et al.; 14 therefore, it seems to be a constant finding of patients with RCAD. On the contrary, it has not been yet described in children with ADPKD or ARPKD.
The limitations of our study are the relative small number of patients, inclusion of children with RCAD and magnesium supplements and its cross-sectional design that could not assess the reproducibility of the data. Despite these limitations, our study is the first study investigating renal magnesium handling in children with ADPKD and only the second study in ARPKD patients.
In conclusion, we could, for the first time, demonstrate that in children with ADPKD hypomagnesaemia is absent and could serve as a marker for differential diagnostic between ADPKD, ARPKD and RCAD in children with cystic kidney diseases of unknown origin where molecular genetic testing for HNF1B and PKD1/PKD2 genes is lacking. On the limited evidence here, it should make clear that while hypomagnesaemia (in the absence of diuretics) appears to rule out ADPKD and ARPKD with accuracy of 0.93, normomagnesaemia does not rule out RCAD at least in those aged less than 3 years and in those on Mg supplements.
Footnotes
Acknowledgements
We thank Jitka Stekrova and Michal Malina for performing the DNA analyses.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Ministry of Health CZ – DRO, University Hospital Motol, Prague, Czech Republic 00064203. Computational resources for R were provided by the MetaCentrum under the LM2010005 programme and CERIT-SC under the Centre CERIT Scientific Cloud programme, part of the Research and development for Innovations Operational Programme, Reg. no. CZ.1.05/3.2.00/08.0144.
Ethical approval
The ethics committee of The Internal Grant Agency of the Czech Ministry of Health (IGA-MZ ČR) approved this study (project number: NT11457–5).
Guarantor
TS.
Contributorship
TS, MP, UJ researched literature and conceived the study. TS, MP, UJ, MF, BS were involved in protocol development, patient recruitment and data analysis. RP, MF ensured Lab Tests and BS realized statistical analysis. TS wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.