
Abstract
The bone remodelling cycle replaces old and damaged bone and is a highly regulated, lifelong process essential for preserving bone integrity and maintaining mineral homeostasis. During the bone remodelling cycle, osteoclastic resorption is tightly coupled to osteoblastic bone formation. The remodelling cycle occurs within the basic multicellular unit and comprises five co-ordinated steps; activation, resorption, reversal, formation and termination. These steps occur simultaneously but asynchronously at multiple different locations within the skeleton. Study of rare human bone disease and animal models have helped to elucidate the cellular and molecular mechanisms that regulate the bone remodelling cycle. The key signalling pathways controlling osteoclastic bone resorption and osteoblastic bone formation are receptor activator of nuclear factor-κB (RANK)/RANK ligand/osteoprotegerin and canonical Wnt signalling. Cytokines, growth factors and prostaglandins act as paracrine regulators of the cycle, whereas endocrine regulators include parathyroid hormone, vitamin D, calcitonin, growth hormone, glucocorticoids, sex hormones, and thyroid hormone. Disruption of the bone remodelling cycle and any resulting imbalance between bone resorption and formation leads to metabolic bone disease, most commonly osteoporosis. The advances in understanding the cellular and molecular mechanisms underlying bone remodelling have also provided targets for pharmacological interventions which include antiresorptive and anabolic therapies. This review will describe the remodelling process and its regulation, discuss osteoporosis and summarize the commonest pharmacological interventions used in its management.
Keywords
Introduction
The skeleton, although perhaps not ordinarily thought of as such, is a dynamic, metabolically active and functionally diverse organ. It provides levers for muscle to allow locomotion, supports and protects vital organs and is the site of haematopoietic marrow. Metabolically, it has roles in both mineral metabolism, via calcium and phosphate homeostasis, and in acid–base balance via buffering hydrogen ions. 1 Recent studies have also suggested that bone may have additional important endocrine roles in fertility, glucose metabolism, appetite regulation and muscle function.2–5
Throughout life, the dynamic skeleton is ‘constructed’ and ‘reconstructed’ by two processes: bone modelling and remodelling. 6 Both processes involve osteoclastic bone resorption and osteoblastic bone formation. In modelling, resorption and formation occur independently at distinct skeletal sites to bring about major changes in bone architecture. By contrast, in remodelling, resorption and formation are tightly coupled both spatially and temporally so that the overall bone volume and structure remain unchanged.
Bone remodelling occurs continuously to repair skeletal damage, prevent accumulation of brittle hyper-mineralized bone and maintain mineral homeostasis by liberating stores of calcium and phosphorus. Small regions of bone are resorbed by osteoclasts and replaced by osteoblasts; this close coordination between resorption and formation ensures that structural integrity is maintained while allowing up to 10% of the skeleton to be replaced each year. 7 Remodelling is regulated by both systemic and local factors and the key signalling pathways have been identified by the study of families with rare bone diseases and in animal models.
This review highlights recent advances in understanding skeletal maintenance and repair and discusses the cellular and molecular mechanisms that underlie the bone remodelling cycle. It emphasizes the central role of the osteocyte in orchestrating both osteoclastic bone resorption and osteoblastic bone formation and describes the key regulatory pathways and drug targets including RANK/RANKL/osteoprotegerin (OPG) and Wnt signalling.
Bone cells
Within bone there are four major skeletal cell types
Cartilage-forming chondrocytes Bone-forming osteoblasts Bone-resorbing osteoclasts Mechanotransducing and regulatory osteocytes
The cellular origin of the skeletal cell types is illustrated in Figure 1, and Table 1 details their structure, function and regulation. Bone lining cells are mature osteoblasts that cover quiescent bone surfaces; however, their role is incompletely understood and they will not be discussed further.
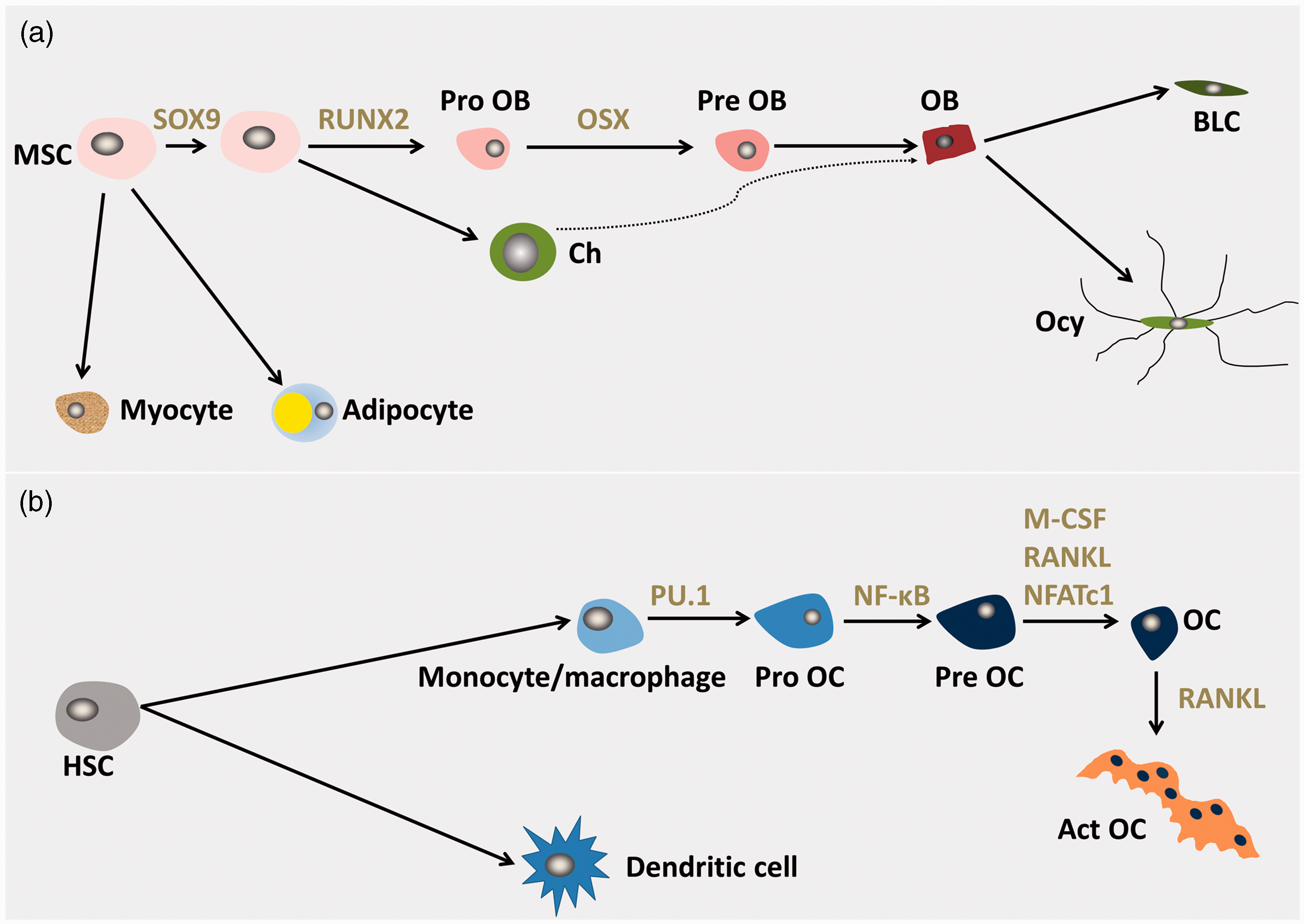
Derivation of bone cells. (a) Mesenchymal stem cells (MSCs) can form adipocytes, chondrocytes (Ch), myocytes or osteoblast precursors (Pro OB), pre-osteoblasts (Pre OB) then osteoblasts (OB). Mature osteoblasts can differentiate into bone lining cells (BLC) or osteocytes (Ocy). Recent evidence suggests that hypertrophic chondrocytes may also differentiate into OBs.10 The key transcriptional regulators in osteoblast differentiation are indicated. Sry-box 9 (SOX9), runt-related transcription factor 2 (Runx2), Osterix (OSX). (b) Haemopoietic stem cells (HSCs), specifically myeloid-committed precursors, differentiate into monocytes/macrophages or dendritic cells. Monocytes/macrophages then differentiate into osteoclast progenitors (Pro OC), pre-osteoclasts (Pre OC) then osteoclasts (OC). Active OC (Act OC) formation is stimulated by RANK ligand.7,20,23,205 The most important cytokines and transcriptional regulators of this pathway are indicated. PU box-binding-1 (PU.1), nuclear factor-κB (NF-κB), macrophage colony-stimulating factor (M-CSF), nuclear factor of activated T cells 1 (NFATc1) and RANKL.
Specialized bone cells involved in the bone remodelling process.

Bone structure
Bone is a combination of osteoid matrix and hydroxyapatite [Ca10(PO4)6(OH)2] crystal but bone also contains water, non-collagenous proteins, lipids and specialized bone cells.1,36
The type 1 collagen bone matrix gives bone elasticity, flexibility and tensile strength. The collagen fibres are made up of three helical chains and combine together to form fibrils. Fibrils are then interwoven and bound by crosslinks. 37 Non-collagenous proteins, adsorbed from the serum, also make up the matrix. The role of such proteins is becoming increasingly clear and their major functions include strengthening the collagen structure and regulating its mineralization. Bone mineral, in the form of hydroxyapatite crystals, is an essential store of calcium and phosphate required for mineral homeostasis and provides the skeleton with mechanical rigidity and compressive strength. Recently, NMR spectroscopy has given new insights into the detailed composition of bone matrix and mineral. 38
Bones fulfil a protective and supportive role but are also essential for locomotion; they are therefore required to be strong yet light. Consequently, bones are made up of two, structurally distinct, types of bone – cortical and trabecular (cancellous). Cortical bone is solid with penetrating vascular canals and makes up the outer dense shell. It has an outer periosteal surface containing blood vessels, nerve endings, osteoblasts and osteoclasts and an inner, endosteal surface adjacent to the marrow. 39 On the endosteal surface of cortical bone is the honeycomb-like trabecular bone, which is made up of a fine network of connecting plates and rods. 8
The structural differences between cortical and trabecular bone underlie their diverse functions. The majority of the mature skeleton (∼80%) is dense cortical bone that has a high torsional resistance and a lower rate of turnover. Nevertheless, it can release mineral in response to a significant or long-lasting deficiency. By contrast, trabecular bone, which is less dense, more elastic, has a higher turnover rate, and high resistance to compression makes up the rest of the skeleton. It serves to provide mechanical support, helping to maintain skeletal strength and integrity with its rods and plates aligned in a pattern that provides maximal strength. Trabecular bone has a large surface area for mineral exchange and is more metabolically active than cortical bone, rapidly liberating minerals in acute insufficiency. 40 Consequently, trabecular bone is also preferentially affected by osteoporosis. 41
The proportions of cortical and trabecular bone present are dependent on the individual bone’s function. In vertebrae, trabecular bone predominates to resist compressive forces. By contrast, long bones, which principally act as levers, are mostly composed of cortical bone to allow them to resist both compressive and torsional forces.41,42
Bone development
The skeleton is formed in two distinct processes. Flat bones such as skull vault are formed by intramembranous ossification where mesenchymal cells differentiate into osteoblasts which secrete and mineralize osteoid directly to form plate-like bones (Figure 2).
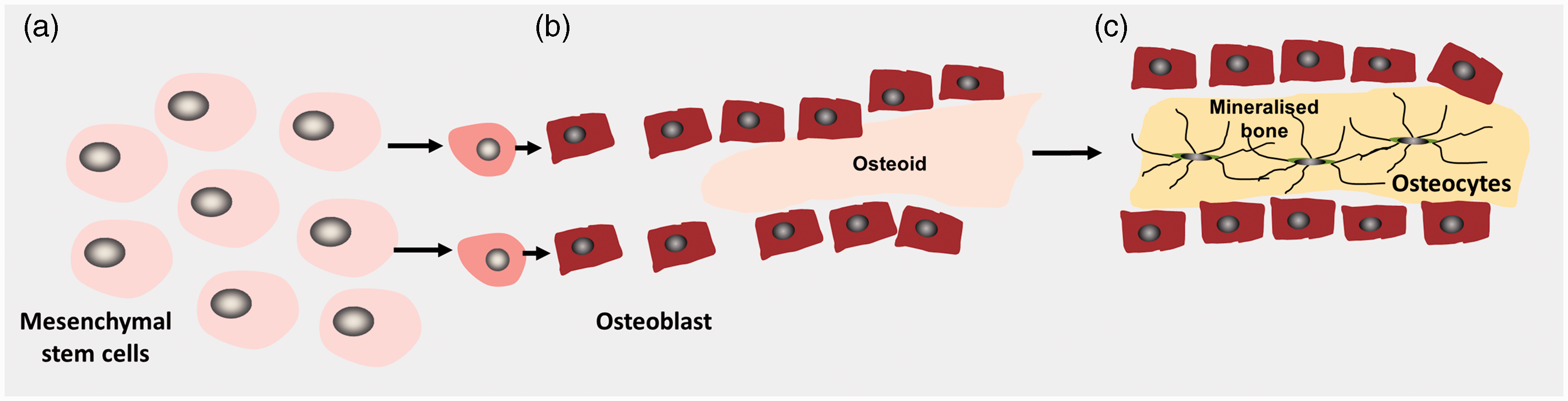
Schematic diagram illustrating intramembranous bone formation. Mesenchymal stem cells differentiate into osteoblasts and form bone directly. (a) Mesenchymal stem cells in connective tissue form a cluster and differentiate in osteoblasts. (b) Mature osteoblasts secrete a type I collagen-rich matrix called osteoid. (c). The osteoid mineralizes to form an ossification centre from which mineralization spreads. Osteoblasts terminally differentiate into osteocytes and become entombed within the newly formed bone matrix.
The multistep process of endochondral bone formation is illustrated in Figure 3. Endochondral ossification forms the majority of the axial and appendicular skeleton. In this process, skeletal elements are initially formed as a cartilage template that is subsequently replaced by bone. Endochondral ossification begins when chondrocytes, differentiate from embryonic mesenchymal stem cells and secrete a collagen II-rich matrix. The chondrocytes proliferate and then subsequently undergo hypertrophic differentiation, secreting a type X collagen-rich matrix which then mineralizes. Chondrocyte apoptosis results in vascularization and formation of the primary ossification centre. The mineralized cartilage acts as a template for subsequent trabecular bone formation mediated by osteoclasts and osteoblasts. Secondary ossification centres also form in the epiphysis at the proximal and distal end of long bones. The chondrocytes that remain between the primary and secondary ossification centres form the growth plate where linear growth occurs until quiescence or fusion at puberty.11,43
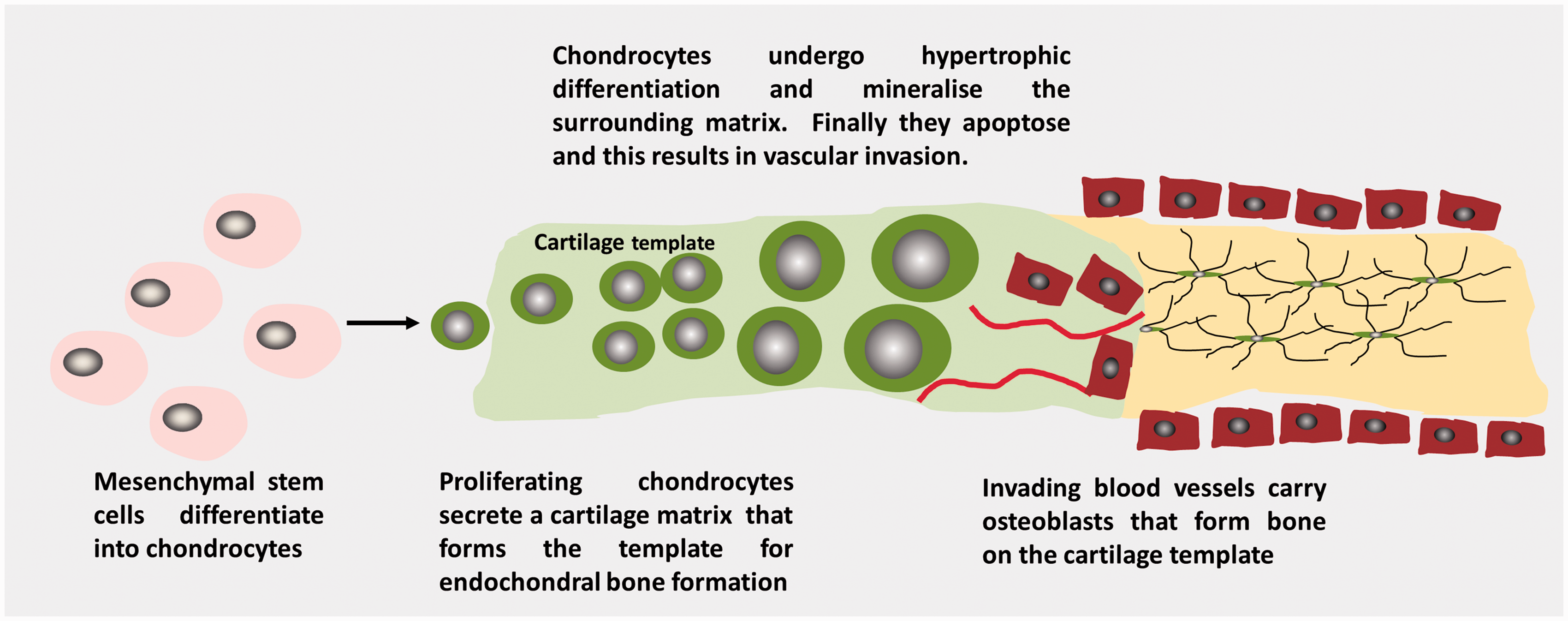
Schematic illustrating endochondral bone formation.
Bone modelling
Bone modelling, which begins early in skeletal development, modifies the size and shape of a bone. In this process, bone resorption and formation must be uncoupled; bone is removed from one anatomical site and new bone is formed at another. One important example of modelling is to preserve skeletal shape during linear growth. In the metaphysis, below the growth plate, there is osteoclastic resorption on the periosteal surface, while there is new bone formation on the inner endosteal surface thus converting the shape of the epiphysis into the diaphysis.44,45When these processes are disrupted, for example following antiresorptive (bisphosphonate) treatment of childhood osteogenesis imperfecta, a dramatic inhibition of normal metaphyseal modelling ‘Metaphyseal inwaisting’ is seen. 46 Modelling is also responsible for radial growth of the diaphysis of long bones. Here, osteoclastic resorption occurs on the endosteal surface, while osteoblastic bone formation occurs at the periosteal surface thus increasing the overall diameter with age.
The majority of bone modelling is completed by skeletal maturity but modelling can still occur even in adulthood such as in an adaptive response to mechanical loading and exercise and in renal bone disease.47–50
Adult bone maintenance
The bone remodelling cycle
The skeleton regulates its own maintenance and repair by remodelling, and this process also provides a mechanism for rapid access to calcium and phosphate to maintain mineral homeostasis.51,52 First defined by Frost, the bone remodelling cycle is a tightly regulated process that replaces old and damaged bone with new. 53 Anatomically, the cycle takes place within a Basic Multicellular Unit (BMU), which is composed of osteoclasts, osteoblasts and a capillary blood supply. 54 The BMU lasts longer than the lifespan of the osteoblasts and osteoclasts within it and so requires constant replenishment of these cells which is critically controlled by the osteocyte. The structure and composition of the BMU vary depending on whether it is located within trabecular or cortical bone. In trabecular bone, the BMU is located on the surface such that a ‘trench’ of bone, called Howship’s lacunae, is resorbed then refilled. By contrast, in cortical bone, the osteoclasts within the BMU form a cutting cone that ‘tunnels’ into the cortex, removing damaged bone. Behind the cutting cone, new bone is then laid down concentrically on the tunnel walls by differentiated osteoblasts to leave a vascular supply within the Haversian canal of the new osteon. 55 In both instances, the BMU is covered by a canopy of cells which delineate the bone remodelling compartment (BRC). The BRC provides a defined area of remodelling with close anatomical coupling of osteoclasts and osteoblasts.56,57
Key steps in the remodelling cycle – Cellular and molecular mechanisms
The remodelling cycle occurs in a highly regulated and stereotyped fashion with five overlapping steps of activation, resorption, reversal, formation and termination occurring over the course of 120–200 days in cortical and trabecular bone, respectively. 58 Osteocytes orchestrate the bone remodelling by regulating osteoclast and osteoblast differentiation and thus bone resorption and formation as per Figure 4.
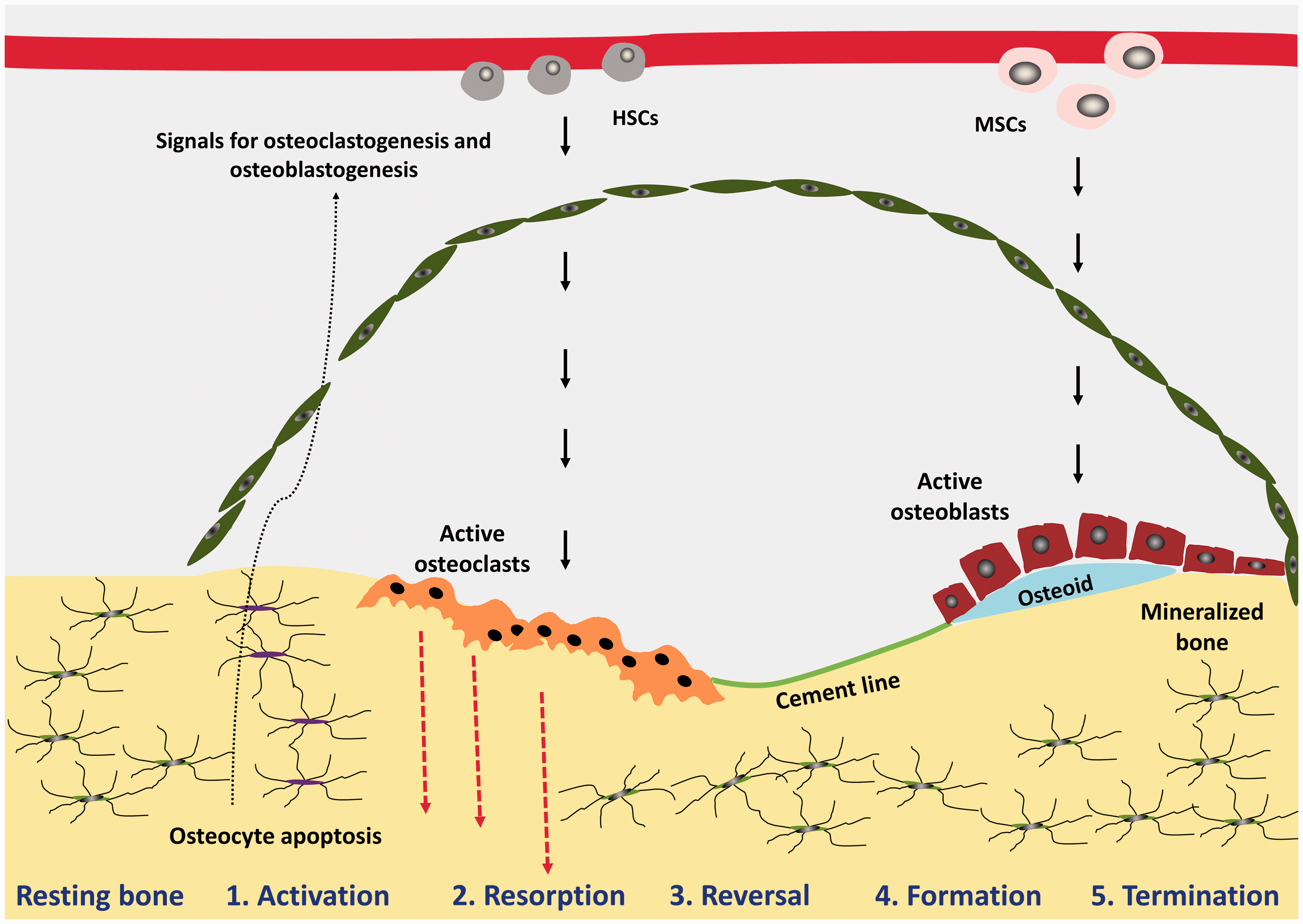
BMU at different phases of the bone remodelling cycle. Schematic diagram of the bone remodelling cycle illustrating the phases of: activation, resorption, reversal, formation and termination. Haemopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
Activation
Osteoclast precursor cells are recruited from the circulation and activated; the bone surface is exposed as the lining cells separate from underlying bone and form a raised canopy over the site to be resorbed. 56 Multiple mononuclear cells fuse to form multinucleated preosteoclasts which bind to the bone matrix to form sealing zones around bone-resorbing compartments, thus isolating the resorption pit from surrounding bone.
Initiation of bone remodelling is the first important step ensuring that, in health, remodelling only takes place when it is required. In targeted remodelling, which refers to removal of a specific area of damaged or old bone, the initiating signal originates from the osteocytes that use their extensive network of dendritic processes to signal to other cells.51,59–62 Osteocyte apoptosis, induced for example by the disruption of osteocyte canaliculi caused by bone matrix microdamage, leads to release of paracrine factors that increase local angiogenesis and recruitment of osteoclast and osteoblast precursors.30,31,60,63 By contrast, non-targeted remodelling refers to remodelling in response to systemic changes in hormones such as parathyroid hormone (PTH), thus allowing access to bone calcium stores and is not directed towards a specific site.
Resorption (approximately two weeks in duration)
Differentiation and activation of osteoclasts are also regulated by osteocytes. Rearrangement of the osteoclast cytoskeleton results in adherence to the bone surface, formation of a sealing zone and generation of a ruffled border that provides a greatly enhanced secretory surface area. Initially, osteoclasts pump protons, generated by Carbonic Anhydrase II, into the resorbing compartment to dissolve the bone mineral. Specifically, the H+-ATPase pumps H+ into lacunae; this is coupled to Cl− transport via a chloride channel thus maintaining electroneutrality. 64 Subsequently, the collagen-rich bone matrix is degraded by proteases such as cathepsin K and matrix metalloproteinases.65,66 The resorption phase is terminated by osteoclasts programmed cell death, ensuring that excess resorption does not occur. 67
Reversal (approximately four to five weeks in duration) 68
The reversal phase, where bone resorption switches to formation, is still not well understood. However, there are thought to be two key events occurring. Firstly, the freshly resorbed bone surface is prepared for deposition of new bone matrix and further signalling occurs that couples resorption to formation, ensuring that there is no net bone loss.69,70 Preparation of the bone surface is carried out by cells of an osteoblastic lineage which remove unmineralized collagen matrix, and a non-collagenous mineralized matrix ‘cement-line’ is then deposited to enhance osteoblastic adherence. 71
The exact signal that couples bone resorption to subsequent formation is not yet fully understood. However, it is likely that the cells of the reversal phase are involved in sending or receiving these signals.72–74
It has been postulated that osteoclasts may be the source of the coupling factor, either secreting cytokines such as interleukin 6 (IL-6), or via a regulatory receptor on their surface such as the Ephrin receptor family and their membrane bound ligand, Ephrins, present on osteoblasts. 75 Other signalling pathways may include matrix-derived factors such as BMP-2, transforming growth factor β and insulin-like growth factor.76,77
Formation (approximately four months in duration) 78
New bone formation can be divided into two parts. Firstly, osteoblasts synthesize and secrete a type 1 collagen-rich osteoid matrix. Secondly, osteoblasts play a part in regulating osteoid mineralization. 60
The process of bone mineralization, whereby hydroxyapatite crystals are deposited amongst collagen fibrils, is complex and its regulation is incompletely understood. Control is exerted by systemic regulation of calcium and phosphate concentrations, local concentration of calcium and phosphate within extracellular matrix vesicles and by local inhibitors of mineralization, including pyrophosphate and non-collagenous proteins such as osteopontin. The ratio of inorganic pyrophosphate to phosphate is a critical regulator of mineralization, and the relative activities of tissue non-specific alkaline phosphatase and ectonucleotide pyrophosphatase are the key determinants of this ratio.79–81
Termination
Once mineralization is complete, osteoblasts undergo apoptosis, change into bone-lining cells or become entombed within the bone matrix and terminally differentiate into osteocytes. Osteocytes play a key role in signalling the end of remodelling via secretion of antagonists to osteogenesis, specifically antagonists of the Wnt signalling pathway such as SOST. 28
Major signalling pathways
The remodelling cycle is tightly regulated to achieve balanced resorption and formation. While systemically released factors play a regulatory role, the fact that remodelling occurs at multiple, anatomically distinct sites at the same time indicates that local regulation is critical to achieving this fine balance. Accordingly, two key pathways, RANKL/RANK/OPG and Wnt, transduce systemically and locally produced signals. Their regulatory role in determining the balance and timing of bone resorption and formation within the remodelling cycle makes them potentially important targets for pharmacological interventions in disease states such as osteoporosis.
RANKL/RANK/OPG signalling pathway
Identification of the RANKL/RANK/OPG Signalling Pathway in the 1990s was a crucial breakthrough in understanding the regulation of osteoclastogenesis in the remodelling cycle and provided the pharmacological target for the novel antiresoprtive denosumab. 82
A permissive concentration of M-CSF, which is expressed by osteocytes and osteoblasts and stimulates RANK expression, is required prior to the action of RANKL.83,84
RANKL binding to its receptor, RANK, on osteoclastic precursor cells, drives further osteoclast differentiation and facilitates fusion, activation and survival.85,86 RANKL/RANK binding induces downstream signalling molecules including mitogen-activated protein kinase, tumour necrosis factor (TNF)-receptor-associated factor 6, NF-κB and c-fos and ultimately activation of key transcription factors, including NFATc1, that regulate the expression of osteoclast genes.23,83,84,87,88
While RANKL can be produced by osteoblasts, osteocytes and chondrocytes, it is the osteocytes, within the bone matrix, that sense changes in load and microdamage that are thought to stimulate osteoclastogenesis via production of RANKL at the initiation of the bone remodelling cycle.34,89
OPG, a decoy receptor for RANKL, was identified prior to the discovery of RANK/RANKL. It is secreted by osteoblasts and osteocytes and is able to inhibit osteoclastic bone resorption by binding to RANKL and preventing its binding to RANK.12,34,90 Thus, the RANKL:OPG ratio is key in the regulation of bone resorption, bone mass and skeletal integrity and is modulated by a number of systemic factors (Figure 5).
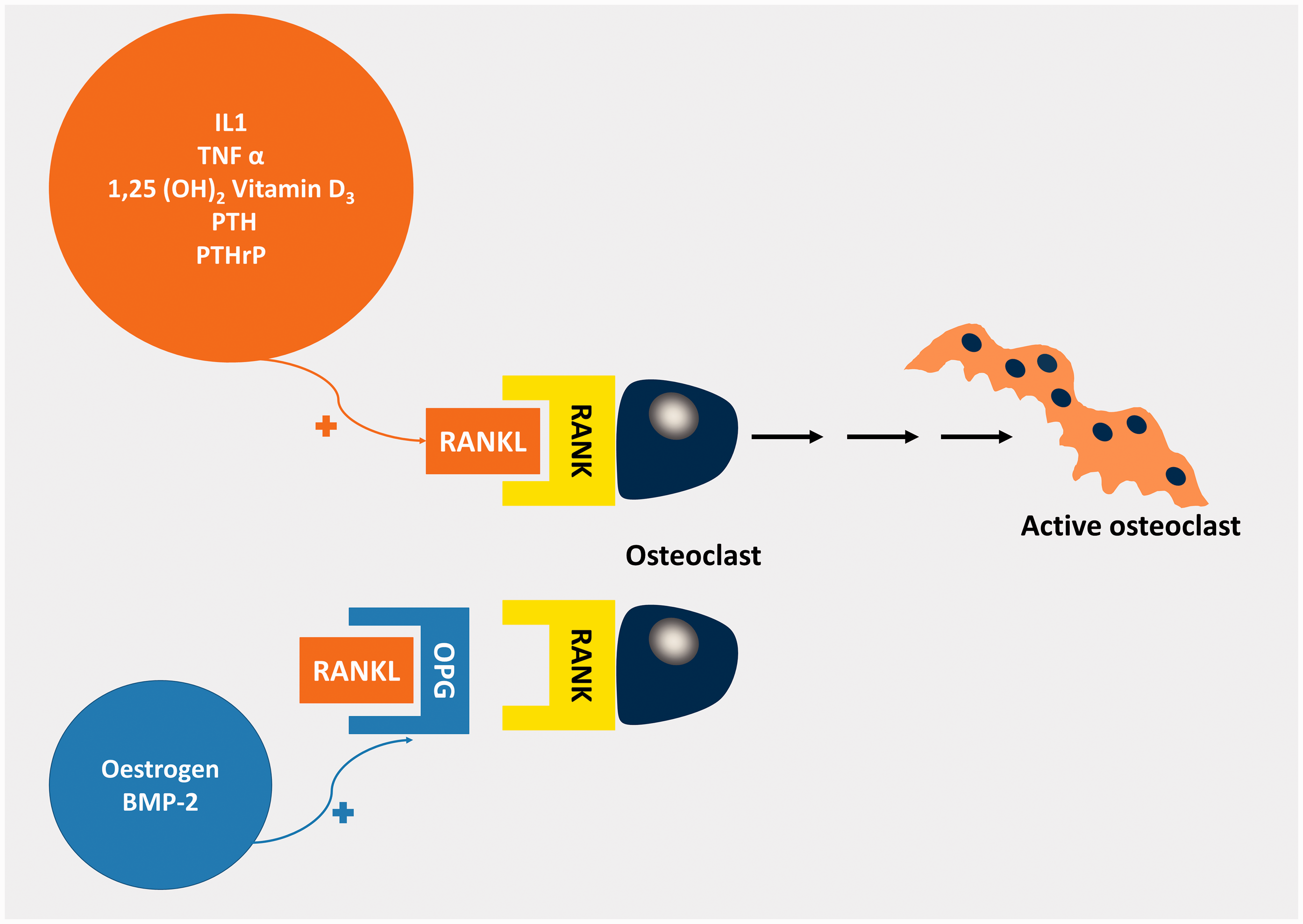
Factors affecting the RANK/RANKL/OPG signalling pathway.206 Oestrogen and Bone morphogenic Protein-2 (BMP-2) induce osteoprotegerin (OPG) expression whereas 1,25(OH)2 Vitamin D3, PTH, PTHrP, IL-1 and tumour necrosis factor α (TNFα) induce RANKL. OPG is a decoy receptor for RANKL blocking its binding to RANK. Thus, it is the RANKL: OPG ratio that determines the rate of osteoclastogenesis.
Wnt signalling
The study of rare human diseases with extreme bone mass phenotypes identified the canonical, β catenin-dependent, Wnt signalling pathway as a major regulator of osteoblastic bone formation (Figure 6).
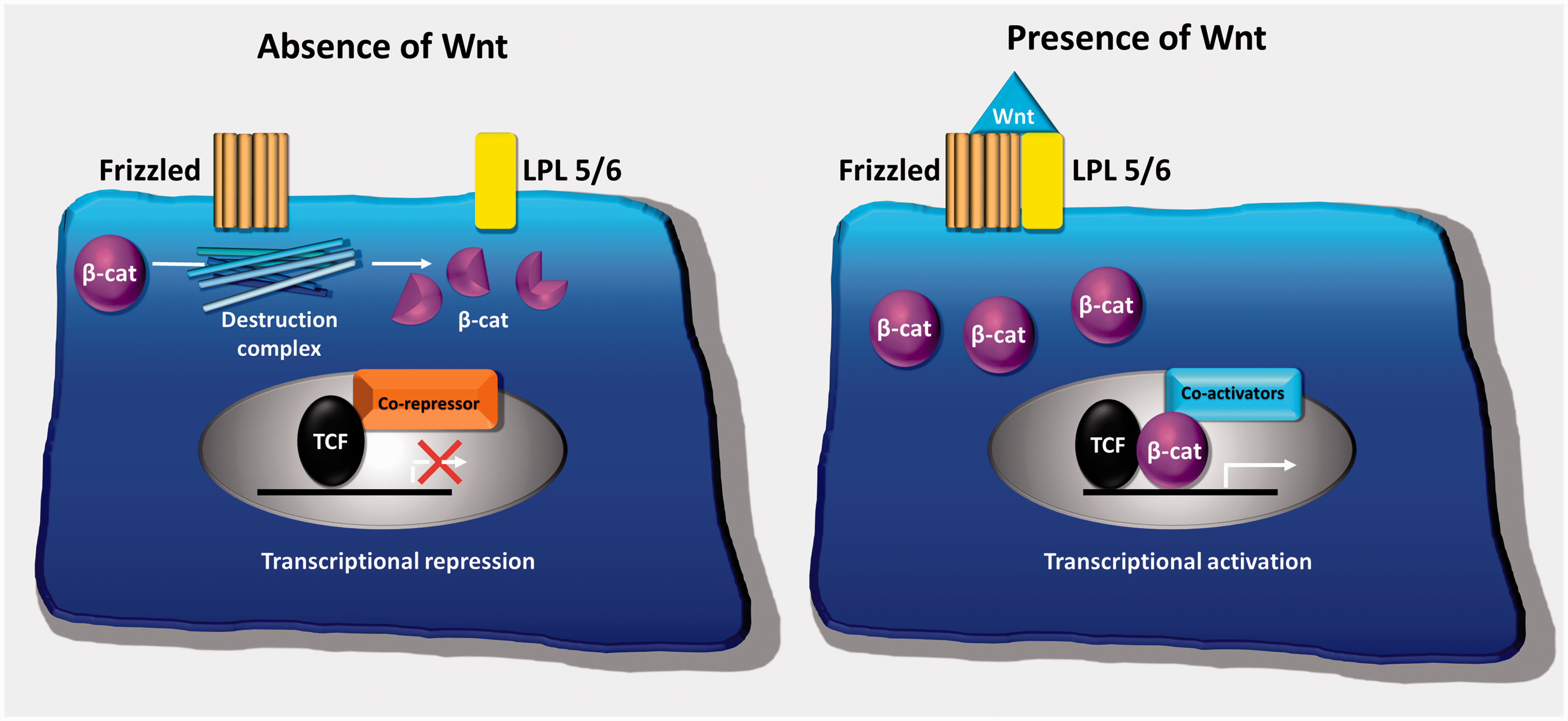
Schematic illustration of canonical Wnt signalling. In the absence of Wnt, Frizzled and its coreceptors LPL5/6 do not interact. The destruction complex, present in the cytoplasm, degrades β-catenin and target gene expression is repressed. In the presence of Wnt, Frizzled binds to its coreceptors and blocks the action of the destruction complex. β-catenin accumulates in the cytoplasm, translocates to the nucleus displacing transcriptional corepressors and recruiting coactivators leading to an increased expression of key target genes involved in osteoblast differentiation.
In the absence of Wnt, a secreted glycoprotein, cytoplasmic β-catenin is targeted for proteosomal degradation by a multisubunit destruction complex which phosphorylates and ubiquitinates β-catenin. Wnt target gene expression is therefore inhibited. When Wnt is present, it binds to a dual receptor complex comprising Frizzled, a seven transmembrane domain receptor, and a coreceptor either lipoprotein-related protein (LPL) 5 or 6. This blocks the action of the destruction complex leading to the accumulation of cytoplasmic β-catenin. The β-catenin then translocates to the nucleus to activate target gene transcription, leading to osteoblast proliferation and differentiation. 91
In patients with osteoporosis-pseudoglioma syndrome, loss of function mutation of the LPL 5 coreceptor results in impaired Wnt signalling and osteoblastic bone formation, resulting in a low bone mass phenotype. 92 The secreted Wnt inhibitor, SOST, was identified by the study of the rare high bone mass disorders, sclerosteosis and Van Buchem disease. These inherited conditions are associated with loss of function mutations of SOST.
SOST is secreted by osteocytes and negatively regulates Wnt signalling by binding the coreceptors LPL 5/6. In quiescent bone, osteocyte expression of the Wnt inhibitors SOST, and DKK-1/2 prevents further bone formation.91,93 However, during the bone remodelling cycle, osteocyte expression of the Wnt-inhibitors declines permitting osteoblastic bone formation to occur after bone resorption. During the termination phase, newly formed osteocytes become entombed within the bone matrix, re-express Wnt inhibitors, resulting in cessation of bone formation. 28
Endocrine regulation of the bone remodelling cycle
PTH
PTH can have directly opposing effects on bone remodelling, depending on duration of exposure. Continuous PTH stimulates bone resorption and is a key physiological mechanism in calcium homeostasis. Furthermore, the prolonged exposure to excess PTH that occurs in primary hyperparathyroidism, due to parathyroid adenoma or parathyroid hyperplasia, results in hypercalcaemia, bone loss and increased fracture risk. 94 Continuous PTH induces both cortical and trabecular bone loss, but cortical bone is more severely affected. These catabolic effects are due to PTH’s modulation of the OPG-RANKL-RANK signalling system. Via action in osteocytes and osteoblasts, continuous PTH increases RANKL and inhibits OPG to stimulate osteoclastogenesis. 95 Monocyte chemoattractant protein 1, which is involved in the recruitment and differentiation of osteoclast precursors, is also increased in response to excess PTH and is thought to play a role in patients with primary hyperparathyroidism. 96
By contrast, intermittently administered PTH is used as an anabolic agent in the treatment of osteoporosis. Intermittent PTH receptor stimulation enhances bone formation via modulation of Wnt signalling. Intermittent PTH signalling reduces the expression of osteocyte-derived Wnt inhibitors SOST and DKK-1, while also increasing the Wnt ligand Wnt10b. The increase in canonical Wnt signalling results in increased osteoblastogenesis, target gene expression and enhanced bone formation.95,97–99
Vitamin D
1,25(OH)2Vitamin D regulates intestinal calcium and phosphate absorption providing the substrates for bone mineralization. However, the physiological actions of 1,25(OH)2Vitamin D in the bone remodelling cycle remain uncertain.
Several studies have reported expression of the vitamin D receptor (VDR) in osteoclast and osteoblast precursors, and in osteocytes, suggesting that vitamin D may also mediate direct effects in bone. VDR expression has been shown in human osteoclast precursors but studies in the mature osteoclast have been contradictory.100–102 Similarly, osteoblast precursors express the VDR, whereas only low levels are detectable in mature osteoblasts.103,104 Despite this, studies in osteocytes have demonstrated VDR expression. 105 Furthermore, in vitro studies have shown activity of the vitamin D-activating enzyme 1α hydroxylase in human osteoblast, osteoclast and mRNA expression in osteocytes suggesting possible local regulation of vitamin D activity in skeletal cells.105–107
By contrast, initial studies in global VDR-deficient mice showed that their abnormal skeletal phenotype could be rescued by dietary calcium supplementation alone, suggesting any direct actions of vitamin D in skeletal cells are likely be limited.108,109 Consistent with this, cell-specific deletion of the VDR in the late osteoblast/osteocyte lineage, using Dmp1-Cre, resulted in no significant skeletal phenotype when animals were fed a normal diet. Nevertheless, these mice were partially resistant to hypercalcaemia and hypomineralization induced by high dose 1,25(OH)2vitamin D, indicating a potential role for the osteoblast VDR in regulating mineralization. 110 Furthermore, osteoblast-specific VDR deletion, using the Col1a1-Cre, resulted in a small increase in trabecular bone volume in older animals 111 while transgenic osteoblast-specific VDR over-expression increased bone mass and strength due to increased osteoblastic bone formation and reduced osteoclastic resorption.112,113
Taken together, these data confirm a primary role for the intestinal VDR in regulating the calcium supply for skeletal mineralization, but suggest that vitamin D may also have direct actions in skeletal cells.
Calcitonin
Calcitonin is synthesized in the parafollicular C-cells of the thyroid, but its physiological role remains uncertain. At pharmacological concentrations, calcitonin inhibits bone resorption, acting via the calcitonin receptor in osteoclasts, to reduce osteoclast number, secretory activity and ruffled border formation.114,115 By contrast, calcitonin-deficient mice show increased bone formation, and at physiological concentrations, calcitonin inhibits the actions of sphingosine-1-phosphate, a coupling factor that links bone formation to resorption.116,117
Thyroid hormone
Thyrotoxicosis is an established cause of secondary osteoporosis and is associated with both increased osteoblastic bone formation and increased osteoclastic bone resorption. Thyroid hormones directly stimulate osteoblast differentiation and mineralization, but it remains uncertain if thyroid hormones have direct action in osteoclasts.
Thyroid hormone deficiency leads to a lengthening of the bone remodelling cycle with low bone turnover and increased bone mass. Conversely, hyperthyroidism increases bone turnover, decreases the duration of the bone remodelling cycle and leads to uncoupling of osteoblastic and osteoclastic activity, resulting in a 10% loss of bone per remodelling cycle. 118
Growth hormone and insulin-like growth factor 1
Growth hormone (GH) induces insulin-like growth factor 1 expression, increasing bone turnover by stimulating both osteoblastic bone formation and osteoclastic bone resorption. Nevertheless, osteoblastic bone formation predominates, leading to a small net increase in bone mass.119,120 By contrast, in GH deficiency, bone resorption outweighs bone formation, ultimately leading to osteoporosis.
Glucocorticoids
At supra-physiological doses, glucocorticoids cause osteoporosis (Table 2). Glucocorticoids inhibit osteoblast differentiation and function and increase osteoblast apoptosis. 121 By contrast, glucocorticoids increase osteoclastic bone resorption by reducing OPG and increasing RANKL expression by osteoblasts and increasing RANK expression in osteoclasts. However, the enhanced bone resorption is only transient and prolonged glucocorticoid treatment results in reduced osteoclast numbers and resorption.122–124 At physiological concentrations, however, glucocorticoids have been shown to have an anabolic effect on bone turnover. 125
Pathophysiology of commonest causes of osteoporosis.
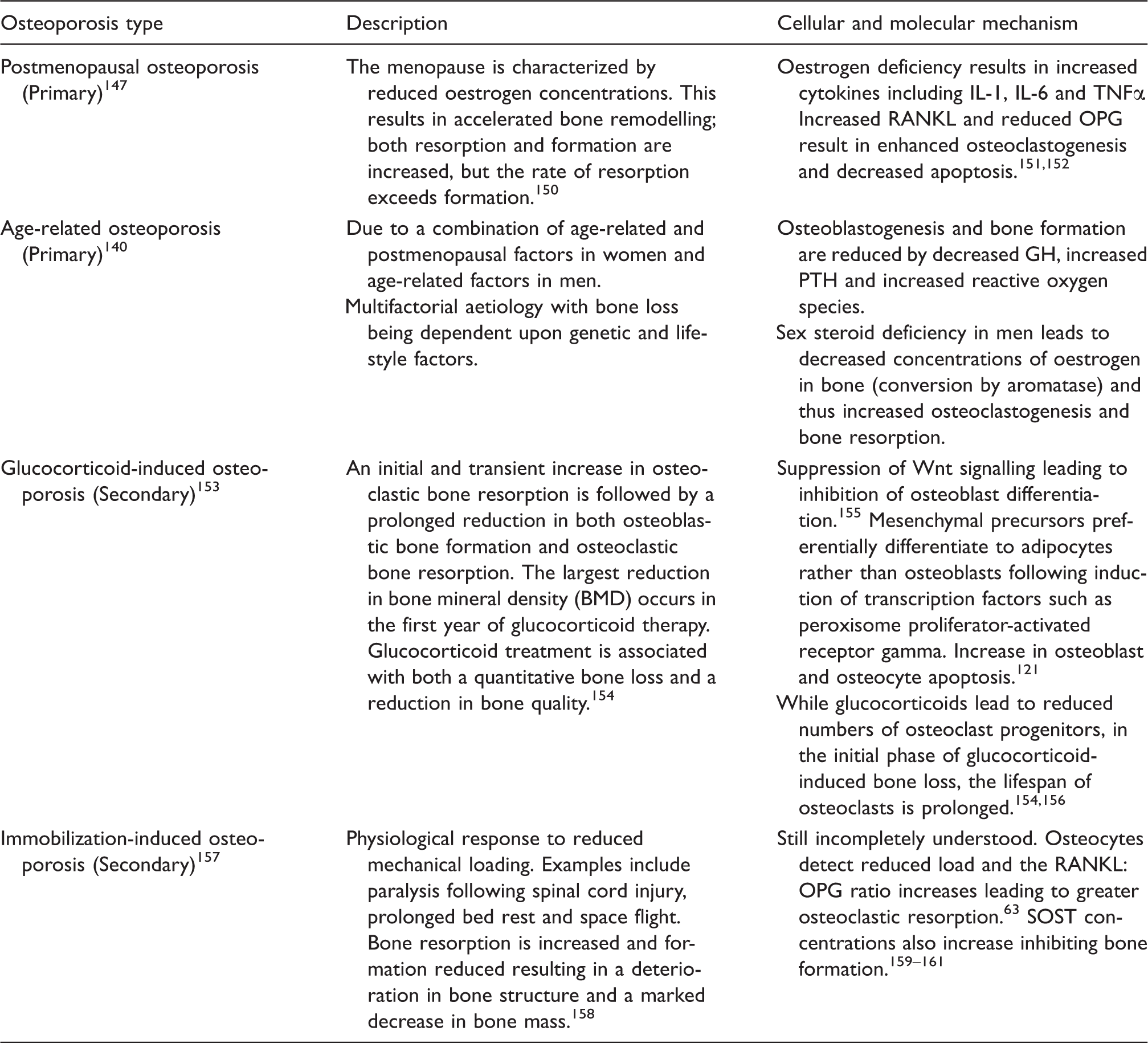
IL: interleukin; TNFα: tumour necrosis factor α; RANKL: receptor activator of nuclear factor ligand; GH: growth hormone; PTH: parathyroid hormone; OPG: osteoprotegerin; SOST: sclerostin.
Sex hormones
Postmenopausal osteoporosis is characterized by uncoupling of the bone remodelling cycle with increased osteoclastic bone resorption relative to osteoblastic bone formation, resulting in net bone loss. Accordingly, oestrogen, acting via the oestrogen receptor-α, inhibits bone resorption by reducing osteoclast number and activity and increasing osteoclast apoptosis. 126 Oestrogen also inhibits osteoblast and osteocyte apoptosis to maintain bone formation and limit bone remodelling.127,128
Aromatase converts androgens to oestrogens, and in postmenopausal women, adrenal steroids are the only source of oestrogens. 129 Thus, women on aromatase inhibitors or with reduced aromatase activity are at an increased risk of osteoporosis. Similarly, aromatase plays an important role in bone mass in men. It has been shown that oestrogen, rather than androgen concentrations, determines bone mass in the aging male population. 130
Androgens, like oestrogens, favour net bone formation by stimulating bone formation and inhibiting resorption. 131 Low levels in men lead to an increased rate of remodelling, which is also due to less oestrogen being aromatized from testosterone.
Oestrogen or androgen deficiency leads to an increase in bone remodelling. While both osteoblastic bone formation and osteoclastic bone resorption are increased, uncoupling results in resorption outweighing formation. 132
Paracrine regulation of the bone remodelling cycle
Growth factors
Transforming growth factor β (TGF β) and BMPs are both members of the TGF β superfamily and are present in the bone matrix. They signal through canonical (Smad) and non-canonical (Smad-independent) pathways. They induce expression of the master osteoblast transcription factor, Runx 2, which is required for initiation of osteoblast differentiation. 133 TGF β1 has also been implicated in coupling of resorption to bone formation by inducing migration of mesenchymal stem cells to resorptive sites. 134
Prostaglandins
Prostaglandins act locally via multiple G-protein coupled receptors to regulate bone resorption and formation. Nevertheless, the exact role of prostaglandins in the bone remodelling cycle remains unclear. For example, prostaglandin E2 (PGE2) is a potent stimulator of bone resorption and is thought to act by increasing the RANKL/OPG ratio to enhance osteoclastogenesis. However, PGE2 also stimulates osteoblast proliferation and differentiation to increase bone formation. It is thought the divergent actions result from PGE2 acting via different G-protein receptors and secondary messenger pathways.135,136
Cytokines
Cytokines, such as IL-1 and IL-6, and TNFα can stimulate osteoclastogenesis, whereas others, such as IL-4 and gamma interferon, inhibit osteoclast formation.137,138
In postmenopausal women, these cytokines play an important role in the pathophysiology of osteoporosis. Oestrogen deficiency results in an increase in IL-1, IL-6 and TNFα, leading to an increased RANKL expression and increased osteoclastogenesis and bone resorption. 139
Abnormalities of the bone remodelling cycle
Osteoporosis
In the bones of healthy adults, the remodelling cycle displays tight coupling between bone resorption and bone formation. Accordingly, several metabolic bone diseases including osteoporosis, hyperparathyroidism, Paget’s disease and osteopetrosis are characterized by loss of such coupling. This field has been previously extensively reviewed by Feng and McDonald, and therefore this review will focus specifically on osteoporosis. 140
Osteoporosis is the most common metabolic bone disorder and resultant fragility fractures are associated with increased morbidity and mortality; its European prevalence is 27.6 million and 1 in 3 women and 1 in 5 men over 50 will sustain osteoporotic fractures.141–143 Osteoporosis may be diagnosed following a fragility fracture or by Dual Energy X-ray Absorptiometry (DEXA) T-score ≤−2.5 (T-score represents the number of standard deviations from the mean of an appropriate young reference population). It may also be suggested by the results of plain radiographs or computed tomography scans. Alternatively, osteoporosis may be defined qualitatively as a decrease in bone mass and strength, leading to increased fracture risk.144,145 Osteoporosis may be a consequence of (i) a failure to reach normal peak bone mass during growth, (ii) a relative increase in bone resorption during adulthood or (iii) a relative reduction in bone formation during adulthood.
Primary osteoporosis is the most common form of osteoporosis and includes both postmenopausal and age-related osteoporosis. By contrast, secondary osteoporosis is a consequence of systemic disease or pharmacological intervention and its aetiology includes:
Endocrine disorders (acromegaly, adrenal insufficiency, Cushing’s syndrome, diabetes, hyperthyroidism, hyperparathyroidism, hyperprolactinaemia, hypogonadism, eating disorders and endometriosis). Connective tissue disease, e.g. rheumatoid arthritis and ankylosing spondylitis. Genetic diseases, including osteogenesis imperfecta, homocystinura, hypophosphatasia Drugs, including glucocorticoids, antiepileptics, anticoagulants, chemotherapy, gonadotrophic-releasing hormone agonists/antagonists and immunosuppressants. Metabolic disorders, including renal and liver disease. Gastrointestinal and nutritional disorders, e.g. parenteral nutrition, gastrectomy or post-gastric bypass, malabsorption, pancreatic insufficiency, inflammatory bowel disease, coeliac, chronic cholestatic disease, primary biliary cholangitis. Disorders of the bone marrow, e.g. myeloma, pernicious anaemia. Multiple sclerosis, congenital porphyria, chronic obstructive pulmonary disease, idiopathic hypercalciuria, idiopathic scoliosis, calcium deficiency.
The most common causes of secondary osteoporosis are glucocorticoid treatment and immobilization. 146
While osteoporosis has many and diverse causes, uncoupling of the bone remodelling cycle and increased bone resorption relative to formation is a common underlying pathophysiological mechanism. The excess skeletal resorption results in structural deterioration and increased fragility. Microscopically sites of osteoclastic bone resorption are incompletely repaired by newly formed bone, resulting in progressive bone loss and increasing cortical porosity.41,147
Initially, osteoporosis may predominantly affect trabecular bone due to its greater surface area. Nevertheless, cortical bone is also affected and its increasing porosity is associated with an increased fracture risk.148,149
The underlying pathophysiology associated with the commonest forms of osteoporosis is detailed in Table 2.
Pharmacological interventions
Current osteoporosis treatments can be divided into (i) those that inhibit osteoclastic bone resorption, such as bisphosphonates, Selective oEstrogen Receptor Modulators and anti-RANKL antibodies and, (ii) those that increase bone formation including strontium ranelate and human PTH (1–34) (Table 3).
Current pharmacological interventions for osteoporosis and guidelines for their use in primary and secondary prevention of osteoporotic fractures.
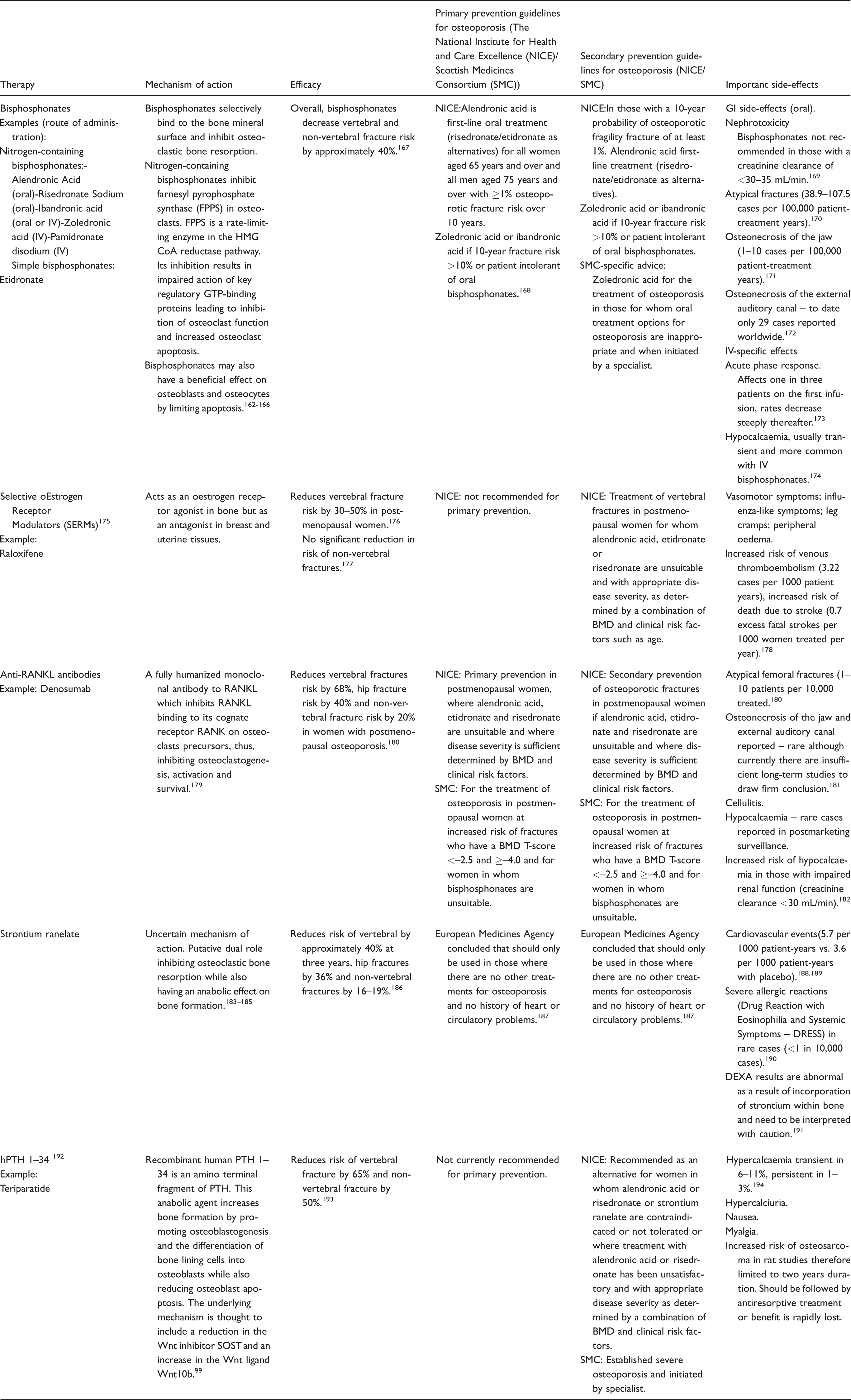
IV: intravenous; RANKL: receptor activator of nuclear factor [κ]-Bligand; HMG CoA: 5-hydroxy-3-methylglutaryl-coenzyme A; GTP: guanosine-5'-triphosphate; PTH: parathyroid hormone; SOST: sclerostin; BMD: bone mineral density.
New osteoporosis treatments
The molecular mechanisms underlying the regulation of the bone remodelling cycle are becoming increasingly well defined and have provided a number of potential therapeutic targets to advance the management of osteoporosis.
Cathepsin K inhibitors (osteoclastic bone resorption)
In an effort to specifically inhibit the resorptive action of osteoclasts, inhibitors of cathepsin K have been developed. Cathepsin K inhibitors impair osteoclastic bone resorption by inhibiting the major protease responsible for Type 1 collagen degradation, the expression of which is restricted predominantly to osteoclasts. However, while several cathepsin K inhibitors have been clinically evaluated, they have not been pursued due to safety concerns. The most promising agent, odanacatib, proved effective, leading to a 72% relative risk reduction in clinical vertebral fractures and a substantial increase in bone mineral density (BMD). 195 However, due to an increased risk of stroke, identified in the phase 3 trial in postmenopausal women, its development was subsequently terminated. 196 Nevertheless, one cathepsin K inhibitor, MIV-711, is still being evaluated in an osteoarthritis clinical trial.
PTH analogues (osteoblastic bone formation)
Abaloparatide is highly selective and high affinity PTHrP analogue which binds to the PTH1 receptor and can be administered subcutaneously or transdermally. In a cohort of 2463 women at high risk of postmenopausal fractures, abaloparatide resulted in an 86% reduction in vertebral and a 43% reduction in non-vertebral fracture. In comparison, daily subcutaneous PTH 1–34 (teriparatide) resulted in an 80% reduction in vertebral and a 30% reduction in non-vertebral fracture. Furthermore, after 18 months of abaloparatide treatment, total hip BMD increased by 3.4% and lumbar spine BMD by 9.2%. 197 The subcutaneous preparation of abaloparatide has now been approved by the USA’s Food and Drug Administration for specified high-risk groups of patients with postmenopausal osteoporosis.
Teriparatide is currently licensed for daily subcutaneous administration. However, a phase 3 trial of once weekly subcutaneous teriparatide at a dose of 56.5 µg in 578 healthy male patients and postmenopausal women with a prevalent vertebral fracture was as effective as daily treatment at preventing new vertebral fractures. Patient acceptability may be enhanced by the less frequent – once weekly – subcutaneous administration of teriparatide. 198
Anti-sclerostin antibodies (osteoblastic bone formation)
One of the most promising groups of anabolic agents targets the Wnt signalling pathway. Anti-SOST antibodies are currently in preclinical trials of which three are known to be in development: romosozumab, blosozumab and BPS804. Their mode of action is to prevent the inhibitory effects of osteocyte-derived SOST on osteoblastic Wnt signalling and thus to increase osteoblastic bone formation. 199 Targeting SOST is particularly attractive as its expression is predominantly limited to skeletal tissues, whereas alternative Wnt antagonists such as DKK-1 or secreted frizzled-related protein 1 are more widely expressed. A Phase II trial in 492 postmenopausal women with low BMD compared monthly romosozumab to placebo, alendronic acid or teriparatide. After 12 months treatment, lumbar spine BMD increased 11.3% with romosozumab, 4.1% with alendronic acid and 7.1% with teriparatide but fell by 0.1% in the placebo group. 200 Furthermore, vertebral fracture risk was reduced by 73% in the romosozumab group in comparison to placebo. 201 Despite these promising results, a recent phase 3 trial reported an increased rate of cardiovascular events in those taking romosozumab in comparison to alendronic acid; therefore, further safety information will be required before it can be considered again for approval.202,203 Interestingly, a recent proteomic analysis in human aortic tissues demonstrated extra-skeletal SOST expression. 204
Summary and conclusions
To preserve its essential load bearing, protective and homeostatic functions, the skeleton must undergo continual remodelling and repair. The bone remodelling cycle ensures that old or damaged bone is replaced and that mineral homeostasis is maintained. Bone remodelling is a highly regulated and stereotyped process characterized by osteoclastic bone resorption followed by osteoblastic bone formation. These two processes are tightly coupled to ensure that bone mass is ultimately preserved.
The osteocyte is the key orchestrator of the bone remodelling cycle. These long-lived, terminally differentiated osteoblasts are entombed within the bone matrix, connected by an extensive dendritic network and act as the skeletal mechanosensor. They respond to microdamage and changes in loading by initiating bone remodelling, and once the repair is complete, they inhibit further bone resorption and formation to maintain bone mass. Furthermore, osteocytes also secrete FGF23, respond to hormones such as PTH to initiate bone resorption and thus maintain mineral homeostasis.
Key osteocyte signalling pathways, including RANK/RANKL/OPG and Wnt, regulate osteoclast and osteoblast differentiation and function and are also the mechanism by which several hormones ultimately exert their actions. Skeletal diseases are frequently associated with dysregulation of the bone remodelling cycle, and the study of rare, inherited metabolic bone diseases has greatly enhanced our understanding of the cellular and molecular mechanisms underlying its regulation. Importantly, these studies have also identified novel therapeutic targets for the prevention and treatment of osteoporosis and other metabolic bone diseases.
Footnotes
Acknowledgement
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JSK is an NIHR Academic Clinical Fellow.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: JHDB is funded by a Wellcome Trust Strategic Award (101123), a Wellcome Trust Joint Investigator Award (110141), a European Commission Horizon, 2020 Grant (666869, THYRAGE) and an MRC Research Grant (N01121X).
Ethical approval
Not applicable.
Guarantor
JHDB.
Contributorship
JSK drafted the article. JHDB provided critical revisions of the article. Both authors reviewed and edited the manuscript and approved the final version of the manuscript.