
Abstract
In the last decade, a large number of neuronal cell-surface antibodies have been described which are responsible for a range of neuroimmunological central nervous system disorders. Unlike the paraneoplastic antibodies which target intracellular antigens, these antibodies appear to be pathogenic and hence identification and prompt treatment can make a substantial impact on clinical outcomes of these patients. We review the common antibodies against the ionotropic glutamate receptors (NMDAR, AMPAR), metabotropic glutamate receptors (mGluR1 and mGluR5), voltage-gated potassium channel-complex proteins (LGI1, CASPR2), and other antibodies targeted against glycine receptor, glutamic acid decarboxylase, gamma-amino butyric acid B, dopamine-2-receptor and dipeptidyl-peptidase-like protein 6.
Keywords
Introduction
The aim of this article is to review a range of newer autoimmune disease markers of the central nervous system (CNS) that can be utilized in assisting clinical diagnosis. These autoantibodies may be paraneoplastic (associated with remote malignancy), non-paraneoplastic (which may be coincidently linked to cancer) or idiopathic in aetiology. The autoantibodies help to diagnose syndromes which often differ from other systemic inflammatory processes, such as demyelination (multiple sclerosis) and acute disseminated encephalomyelitis where there may be no neuronal specific biomarkers. 1
A range of novel autoantibodies have been identified that exert their effects by interfering with the cell-to-cell signal transmission process. 2 It is pertinent to first briefly appreciate the mechanisms involved in the neuronal signal transmission that may be vulnerable to the interference by the neuronal autoimmune markers. Signal transmission is a complex subject, 3 and a detailed discussion is beyond the scope of this forum, but attempts will be made to simplify it wherever necessary in order to assist the reader to comprehend the role of these autoantibodies in the neurological diseases. We will attempt to restrict the discussion mainly to antibodies relevant to the CNS with limbic encephalitis (LE) as the prototype, since the peripheral nervous system autoimmunity is an extensive topic in itself. 4 Other antibodies which cause CNS disease like those against aquaporin-4/myelin oligodendrocyte glycoprotein (neuromyelitis optica) and gangliosides (Miller Fisher syndrome and Bickerstaff's encephalitis) will also be not covered in this review, since their pathogenetic mechanisms are marginally different.5,6
Neuronal signal transmission
For in-depth information on this topic, readers are directed to reviews by Rousseaux 7 and Bowie. 8 In brief, electrical signal (action potential) travelling along the axonal fibre reaches an interconnecting junction (synapse) between two neurons where it is converted into a chemical form (majority of synapses in the nervous system are chemical) for continuation onto the next neuron and ultimately the target area. 9 This is the elementary process for cell-to-cell communication in the nervous system (similar mechanism exists for neuromuscular transmission as well, where the synapse is between the nerve and the muscle).
The arrival of electrical impulse at the synapse instigates the influx of calcium ions through the voltage-gated calcium channels, located in the membrane of the presynaptic terminal (bouton). Consequently, calcium influx prompts synaptic vesicles containing neurotransmitter (chemical signal) to migrate and dock at the presynaptic membrane in preparation for release into the synaptic cleft. Once discharged, vesicular content diffuses across to the postsynaptic membrane where it binds to its target protein, known as a receptor (Figure 1). The ensuing cascade of events and the final response resulting from activation of the postsynaptic membrane receptor depend on the types of neurotransmitter released. The response can be either another electrical signal or activation of a secondary messenger system, resulting in either excitation or inhibition of the postsynaptic cell.
Neuronal signal – ionotropic transmission.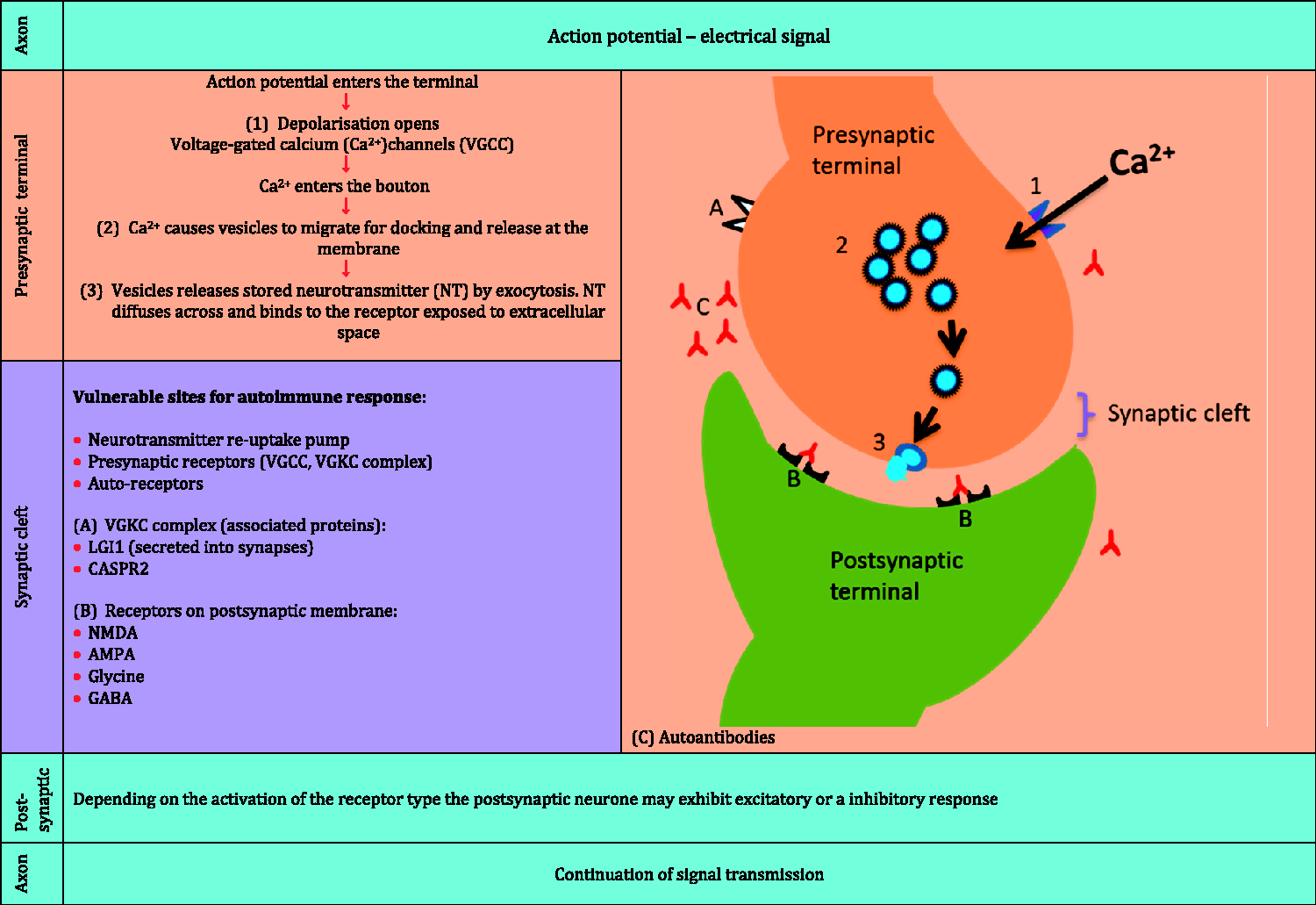
In the CNS, the excitatory neurotransmitter glutamate is responsible for around 50% of the cell-to-cell communication across the synapses.
7
It is also a precursor for the inhibitory neurotransmitter known as gamma-amino butyric acid (GABA) whose production is catalysed by glutamic acid decarboxylase (GAD). A summary of glutamate metabolism (synthesis, packaging, release and removal) is shown in Figure 2. Glutamate is generated from the metabolites of the Kreb’s cycle and packaged in the vesicles, waiting for a signal to be released from the presynaptic terminals. Once released, glutamate can bind with two types of receptors with distinct molecular identity. First type is designated ionotropic glutamate receptor (iGluR) as its binding opens the ion channels found mainly on the postsynaptic terminals. These are considered as being responsible for a number of CNS disorders. These are distinct subtypes of glutamate receptors which have been named after and characterized by using high affinity pharmacological agonists that specifically activate each type and include N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainic acid (kainate).The iGluR type, crucial to the present discussion, is a transmembrane pore (an ion channel) which on activation by glutamate promotes calcium and sodium ion influx into the postsynaptic cell.
Glutamate metabolism.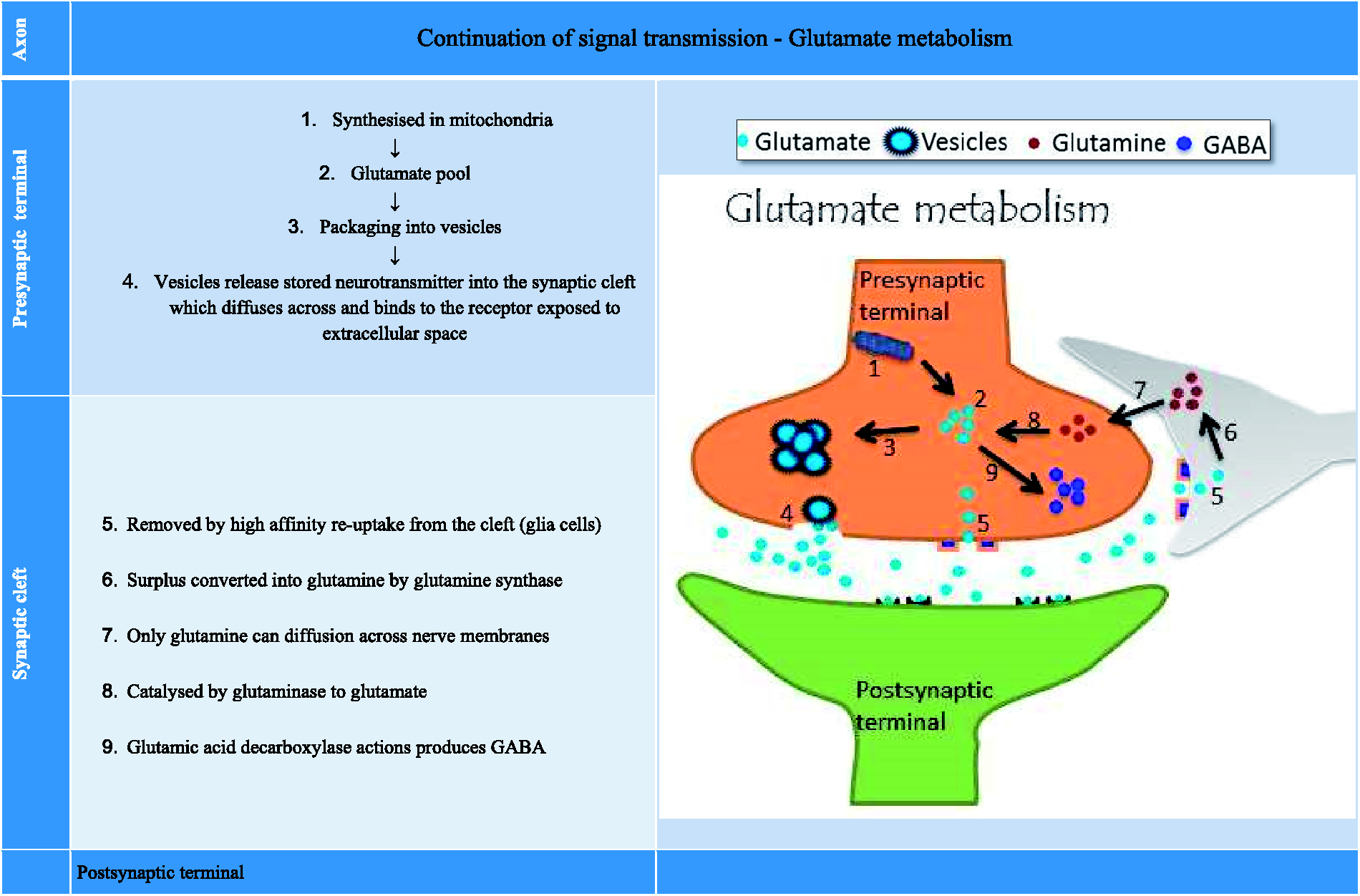
In general, the duration of signal is determined by how long the neurotransmitters remain available in the synaptic cleft (including glutamate) and their actions are terminated by high affinity re-uptake mechanisms located on the presynaptic membrane, thus recycling/replenishing the intracellular supply.
Glutamate can bind to a second type of membrane proteins referred to as metabotropic glutamate receptors (mGluR). These are frequently located on the postsynaptic membrane and produce alterations in intracellular messengers (G-protein), thus resulting in slower response that can either increase or decrease the excitability of the postsynaptic neuron. For further details, see reviews by Rousseaux 7 and Bowie. 8
Every step in the synaptic transmission and its regulation is vulnerable and is a potential target for autoimmune dysregulation, leading to disruption in normal communication and malfunction of the neuronal pathway, manifesting in neurological symptoms.
CNS autoimmunity
In simplistic terms, autoimmune disease is characterized by the activation of the immune system against self-antigens, which is in contrast to the inherent ability of the organism to avoid responsiveness to self-antigens, a process known as tolerance. This inherited ability of the organism serves to eliminate and/or curtail non-self-entities (such as viruses or bacteria) that are likely to compromise health. Irrespective of the reasons for the breakdown in tolerance, the immune system attacks the self-antigens in a sustained and persistent manner, leading to autoimmune disease. This is accompanied by evidence of T- and/or B-cell-mediated response, which is observed as infiltration at the affected site and production of intrathecal immunoglobulin in the CNS and/or the presence of specific circulating antibodies, underscoring the inflammatory contributions of the immune system to the disease process. 10
Once the autoimmune reaction is initiated, it is self-perpetuating and the ongoing tissue damage disrupts the delicate balance between health and disease, in favour of the latter. Thus, tissues, organs and multiple systems harbouring autoantigens, including the nervous system, are targeted relentlessly with potentially fatal consequences. Regardless of the precise mechanism of immune-mediated destruction of the neurons, the resulting neuroimmunological disorder produces a broad spectrum of clinical conditions that can affect both the central and peripheral nervous system. 1 Among the clinical disorders affecting the CNS, a good proportion of the abnormal neurology is due to malignancy-associated immune damage to cerebellum followed by the disorders associated with inflammation of the limbic system, 11 leading to a clinical ailment known as LE. Until recently, this condition has been considered as being mainly paraneoplastic and often associated with lung cancer 12 but in the recent past, this condition has been recognized as also being non-paraneoplastic and usually responsive to immunotherapy availing a greater opportunity for treatment and recovery. 1
Limbic encephalitis
LE is an inflammatory disease of the limbic system (which comprises of the hippocampus, amygdala, hypothalamus, cingulate cortex, fornix and thalamus). LE is characterized by short-term memory deficits, psychiatric manifestations, abnormalities in the MRI and electrical activity.10,13 Many clinical features are common among both types of LE (paraneoplastic and non-paraneoplastic) and there are some signs and symptoms which are specific for each group. If left untreated, irreversible damage and death can occur. 10
A recent UK-based prospective study of encephalitis have divided its causes into three major categories with the largest being infection (42%) where the offending pathogen in most cases is Herpes simplex virus. For the next largest group, the cause is not known (idiopathic) (37%) and the third group comprises 21% with the aetiology due to an acute immune-mediated process, 11 the latter group includes postviral infection and the newer autoantibodies that contribute to the already existing list of classical paraneoplastic neurological antibodies such as Hu (ANNA1), Ma, CV2/CRMP5 and amphiphysin, which can also be associated with LE. It is entirely possible that a substantial proportion of patients who are now classified as ‘idiopathic’ may turn out to have an autoimmune aetiology.
LE can involve one of several antigenic targets (receptors) associated with glutamic acid (glutamate), GABA or glycine transmission. There are also other sensitive destinations which include protein networks associated with VGKCs (Leucine-rich glioma inactivated protein 1 (LGI1), Contactin-associated protein 2 (CASPR2) and Contactin-2).
Classical diagnostic markers
The classical, paraneoplastic neuronal antibodies are considered non-pathogenic, with their target binding sites located inside the cells, and are referred to as ‘intracellular acting’. They are thought to exert their destructive effects on the neurones through cytotoxic T-cell intervention. 14 These paraneoplastic neuronal antibodies are very specific for the disease of the nervous system and usually indicate the presence of an underlying peripheral malignancy which may not be detectable at the time of presentation, thus warranting the follow-up of the patient for up to five years. 15 Treatment of the underlying tumour is paramount in disorders associated with these antibodies. However, unlike the newer cell-surface antibodies, removal of the classical paraneoplastic antibodies using plasma exchange, etc. is unlikely to be clinically meaningful.
New diagnostic markers
In contrast, the mode of action of the newer pathogenic autoantibodies differs from that of the classical paraneoplastic neurological antibodies in that they have their antigens located on the pre- or postsynaptic junction membranes. 14 These antigenic sites are exposed to the extracellular environment for providing an easy access to both the autoimmune system and therapeutic intervention. During the interaction of the autoantibodies with these extracellular antigens, the propagation of signal transmission along the nerve fibre is impaired, precipitating abnormal neuropsychiatric function. This can sometimes lead to a medical emergency, requiring urgent and accurate diagnosis to prevent significant morbidity and mortality.
These autoantibodies have been grouped as ‘neuronal surface acting’ with immune-mediated pathology often with intrathecal synthesis and successful clinical and immunological response to immunomodulatory therapy. 14 There is a full repertoire of these autoantibodies available routinely in a few specialist centres including ourselves in Birmingham and consists of the following reactivities: NMDAR, AMPAR1, AMPAR2, dipeptidyl-peptidase-like protein-6 (DPPX), LGI1, CASPR2 and gamma-aminobutyric acid receptor type B (GABABR1).
NMDA receptor antibodies
In recent years, a treatable form of LE linked to the immune response against the extracellular NMDA receptor was discovered. 16 This is a severe type of encephalitis and can manifest with psychotic behaviour with complex symptoms (memory problems, seizures, unresponsiveness, dyskinesias, autonomic instability and hyperventilation) requiring multidisciplinary care. 16 There appear to be two phases of the disease, the early one is associated with cerebrospinal fluid (CSF) lymphocytosis and in the latter phase, the appearance of CSF oligoclonal bands is observed in the majority of patients. The treatment appears to be more effective in the early stages of the disease and initially consists of immunotherapy (steroids, intravenous immunoglobulins and/or plasmapheresis) and tumour removal. 17 Those patients who are non-responders may be treated with rituximab/cyclophosphamide. Initially, NMDAR antibodies were found predominantly in children and young adults with or without underlying cancer linkage. The underlying cancer association has been reported to be age, gender and ethnicity specific 18 with 80% of female population being affected (ovarian teratoma being the most common neoplastic link). 18 The paraneoplastic association is less commonly observed as the disease phenotype expands with increased awareness. Clinically, neuropsychiatric symptoms together with involuntary movements, ocular phenomena and autonomic manifestations can be encountered by the physician. NMDAR antibodies can be produced intrathecally and the levels of antibodies can potentially be higher in the CSF. However, in the vast majority of patients, serum antibodies are positive and serum is often used as the best screening tool. If the phenotype is atypical, CSF antibodies are useful to confirm the diagnosis. 19
NMDA receptor comprises four subunits (two NR1 and two NR2) each with a molecular size of 100 kDa and together these form an ion channel that is housing the binding sites for both glycine (NR1) and glutamate (NR2). The NR1 subunit is antigenic and the anti-NMDAR immunoglobulin G (IgG) binds to it (Figure 3(a)) and cross-links with the receptor to form a complex which rapidly internalises, thus reversibly reducing receptor density on the postsynaptic neurone.
20
As a result of this, neuronal glutamate signal transmission is hampered and this process is responsible for the CNS dysfunction. Similar mode of action of autoantibodies to the NR2A/B subunit is seen in a number of patients with neuropsychiatric lupus.
21
Human serum from patients with autoimmune encephalitis showing staining of relevant cytoplasmic proteins in HEK cells transfected with (a) NMDAR, (b) CASPR2, (c) LGI1 and (d) GABABR1. As you can see the transfection is partial, thus providing an inbuilt negative control.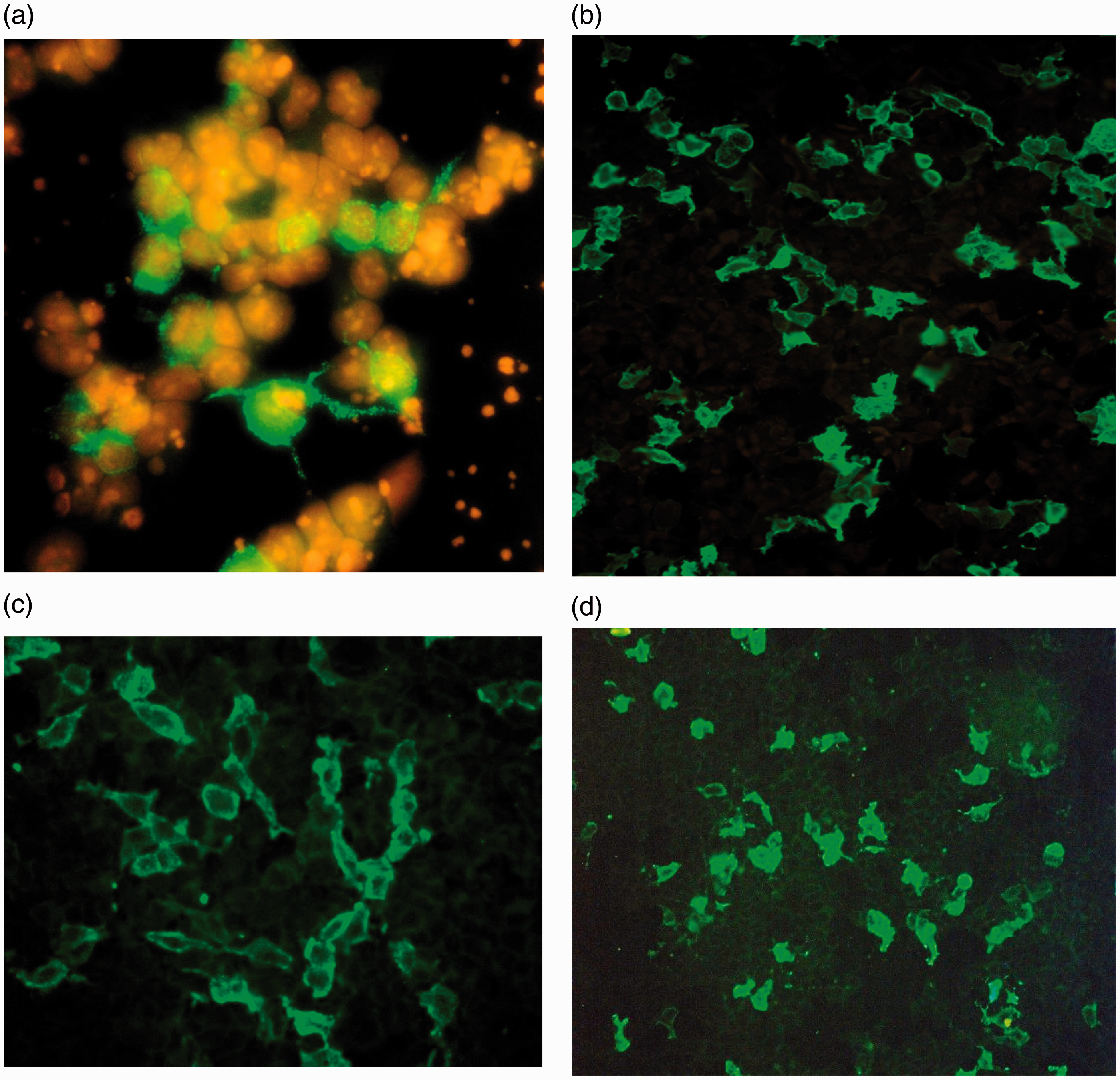
Our experience confirms that NMDAR autoantibody is the most common type of the neuronal surface antibodies found in our cohort of patients with LE, using the fixed transfected cell assay. 22
AMPA receptor antibodies
Antibodies to AMPAR, an ionotropic glutamate receptor, have been described in middle-aged women with classical limbic dysfunction and the majority have an underlying malignancy. AMPAR encephalitis may have psychiatric symptoms on presentation like anti-NMDAR encephalitis. 23 The antibody disrupts excitatory neurotransmission by reacting with the GluR1 and GluR2 subunits of AMPAR and reversibly decreases the synaptic receptor density in a similar manner to the mode of action of NMDAR antibody. The experience with this antibody-associated syndrome is limited as there have only been around 20 or so cases published. Detecting this antibody should lead to a hunt for underlying neoplasm, most commonly lung, breast or thymus. 24
LE associated with AMPAR antibodies is often paraneoplastic (64%) but responds well to treatment, although with frequent relapses.24–26 Antibodies to AMPA-GluR3 antibodies are seen in about 25–30% of patients with autoimmune epilepsies. 21
Antibodies against mGluR
The metabotropic glutamate receptors, coupled to G-protein, are activated by glutamate and can be found in the neuronal synapses at a high concentration in the cerebellum, hippocampus and cerebral cortex.
The antibodies to mGluR1 receptor have been found in the CSF of some patients with paraneoplastic cerebellar degeneration. 26 These receptors can be visualized immunohistochemically on mouse brain sections which show punctuate Purkinje cell staining of the molecular layer of the cerebellum (cerebellum has three layers histologically namely molecular layer, containing the flattened dendritic trees of the Purkinje cells, granule cell layer and the Purkinje cell layer in the middle, which contain the bodies of Purkinje cells) which can be confirmed using a cell-based assay. After injection of anti-mGluR1 antibodies, animal models develop ataxia and eye movement changes. Immunotherapy is considered very efficacious. 21
Similarly, mGluR5 antibodies have been reported albeit very rarely in patients with Hodgkin’s lymphoma and LE (also known as Ophelia syndrome). 27 Unlike the mGluR1 antibodies, mGluR5 antibodies react with the neuropil of the hippocampus and the cell surface of live hippocampal neurons.
GABA antibodies
As mention above, GABA is synthesized from glutamate and catalysed by the action of the enzyme GAD. However, GABA exerts an opposite response to its parent compound, i.e. it is a major inhibitory neurotransmitter in the CNS and activates three types of receptors (known as GABA type A, B and C). The GABABR1 receptors, coupled to a G-protein, are abundantly expressed in the hippocampus, thalamus and cerebellum and its action plays an important part in modulating memory and other cognitive functions. This normal function can be disrupted by the autoimmune processes against GAD enzyme and/or the direct effect on the receptor. In terms of mode of action, GABABR1 antibodies (Figure 3(d)) alter synaptic function without altering the synaptic levels of receptors as seen with NMDAR antibodies. The precise mechanism involved remains to be determined. 28
In a study by Hoftberger et al., 28 almost all of the 20 patients with GABABR1 antibody developed LE with distinct clinical presentation (memory loss, confusion, hallucinations personality changes and seizures) with or without cancer. Fifty per cent of the cases were paraneoplastic and in all the underlying malignancy was small cell lung carcinoma (SCLC), which preceded the neurological deficit. Paraneoplastic LE-associated GABABR1 antibody can coexist with other antibodies such as SRY-related high-mobility-group box (SOX1), amphiphysin or Ri (ANNA2) and when present represents significantly poorer prognosis and survival rate compared to those patients with SCLC and only GABABR1 antibodies.
Clinically, four main clinical syndromes have been reported among the Hoftberger cohort, which include LE with seizures, status epilepticus, ataxia and opsoclonus–myoclonus syndrome. 28 Seizures are often prominent and this is consistent with the extracellular localization of these antibodies.
VGKC-complex antibodies
VGKC is also a transmembrane protein forming channels specific for potassium movement and have a crucial role in returning the depolarized cell harbouring VGKC receptor to a resting state. Up until recently, all the VGKC antibodies analysed with radioimmunoprecipitation assay were thought to be directly against the ion channels 29 but evidence has emerged that the actual target autoantigens are synaptic and axonal neuronal proteins known as LGI1 and CASPR2 which co-precipitate with VGKC were responsible for the positive signal in radioimmunoprecipitation assay. 30 Some patients have antibodies against Contactin (another protein associated with VGKC) and there maybe yet other unknown targets associated with the VGKC-complex proteins. 30
LGI1 antibodies
LGI1, a 60 kDa presynaptic glycoprotein, is essential for normal functioning of the developed synapses and its mutation has been associated with diverse pathologies such as epilepsy, psychiatric disorders and hyponatraemia.
10
It is a secreted protein that forms a bridge between presynaptic
CASPR2 antibodies
CASPR2, a cell-adhesion molecule, is a transmembrane axonal protein belonging to the Neurexin IV family that is thought to organise and concentrate VGKC at the juxtaparanodes of the myelinated axons. 34 The antibodies to CASPR2 target multiple epitopes on the extracellular domain of the protein. 35 Interaction with a particular site on CASPR2 protein may determine the severity of the pathogenic response.
CASPR2 autoimmunity (Figure 3(b)) is less common than LGI1, 29 and affected patients may have encephalitis, acquired neuromyotonia or both, resulting from altered axonal potassium currents. The clinical spectrum can be very similar to patients with LGI1 antibodies but peripheral symptoms like neuromyotonia, neuropathic pain and autonomic dysfunction are relatively more common in CASPR2 antibody positive patients with the onset being either acute or subacute; affecting both genders and manifests around the age of 60 years. 36
DPPX antibodies
DPPX is a cell surface auxiliary subunit of the Kv4.2 of VGKCs in the CNS and is expressed in the hippocampus, cerebellum and myenteric plexus. It has a direct role in the firing patterns of action potentials. 37
The DPPX autoantibody-associated encephalitis often starts with diarrhoea or gastrointestinal dysfunction and patients may develop substantial weight loss. 38 Autoimmunity to the expressed DPPX in the gut may explain these gastrointestinal symptoms which can accompany the onset of brain or brainstem disorder and symptoms of CNS hyperexcitability (seizures, tremors, myoclonus and nystagmus). 39 The autoimmune nature of the illness has been demonstrated by the presence of CSF pleocytosis, oligoclonal bands and neurological response to intensive immunotherapy. 37
Experience from two dozen published cases suggest that this is also a potentially treatable disorder. 40 DPPX is a new addition to the differential diagnosis for acquired neurological disorder and appears to respond well to early initiated immunotherapy with a tendency for patients to relapse. Optimal neurological outcomes may require long-term immunosuppressant therapy. 39
Glycine receptor antibodies
The amino acid, glycine is a neurotransmitter that is involved in the control of motor rhythm generation, coordination of spinal reflexes and processing of sensory signal. In addition, glycine is required for the normal function of NMDA receptors.
Glycine released from presynaptic terminal, mediated by Ca2+, activates the postsynaptic glycine receptor (GlyR). These are ligand-gated pentameric transmembrane ion channels which then mediate inhibitory neurotransmission in postsynaptic cells. 41 GlyR are distributed throughout the CNS but highest densities can be found in the medulla oblongata, pons and spinal cord.
IgG1 complement-fixing antibodies have been identified using α-1 GlyR transfected cells in patients with progressive encephalomyelitis, rigidity and myoclonus (PERM). Patients often have painful muscle spasms triggered by sensory stimuli, frequent falls, excessive startle response (hyperekplexia), autonomic dysfunction and eye movement disorders. 42
Dopamine 2 receptor antibodies
Dopamine (3-hydroxytyramine) is a neurotransmitter that interacts with a family of five membrane receptors coupled to G-protein and are subdivided into two groups based on their ability to promote (D1-like receptor) or inhibit (D2-like receptor) intracellular secondary messenger, cAMP. 43 Among the two groups, Dopamine-2-receptor (D2R) type is of interest, as a target of the immune system, the affected subjects have prominent involuntary movements similar to Parkinsonism, dystonia and chorea and are thought to have basal ganglia involvement. 44 Dale’s cohort group contained mainly children and no tumours were found, but paraneoplastic nature of the disease could not be ruled out. 44
The D2R encephalitis, a subtype often referred to as basal ganglia encephalitis, responds with complete clinical recovery, concomitantly with a fall in serum D2R IgG during early immunotherapy, while a delay in treatment may result is residual neurological and psychiatric sequelae. 45
GAD antibodies
GAD is an intracellular enzyme involved in the conversion of glutamic acid to GABA, an inhibitory neurotransmitter in the brain. GAD exists as two isoforms (65 and 67 kDa) which are expressed in the CNS, pancreatic islet cells, testis, oviduct and ovary. 46 GAD 65 (found in pancreatic beta cells) is 200 times more abundant that GAD 67 (abundantly expressed in the CNS and the pancreatic alpha cells). Antibodies against GAD are usually associated with diabetes mellitus, Stiff Person Syndrome, cerebellar ataxia, epilepsy and non-paraneoplastic LE. 47
Treatment of neurological diseases secondary to cell-surface neuronal antibodies
Irrespective of the type of antibody responsible for the neurological syndrome, most patients respond to immunotherapy. Unless the patient is very sick (e.g. in intensive care or having status epilepticus), most authors claim to start treatment with intravenous steroids followed by oral steroid maintenance therapy. If the symptoms are very severe at the onset or the initial steroid therapy is ineffective, patients might require a course of intravenous immunoglobulin or plasma exchange.17,18 An exhaustive search for any underlying neoplasia should be undertaken (see Algorithm – Figure 4) and if the tumour is found, this needs to be removed for the immunotherapy to be fully effective.17,48 Patients who do not respond to the first-line therapy as above, might require second-line agents in the form of cyclophosphamide or Rituximab (Figure 4).
49
Some patients would require long-term immunosuppressive therapy, especially when the antibodies are directed against NMDAR.
Suggested treatment algorithm for autoimmune encephalitis, with emphasis on the common antibodies, i.e. NMDAR and LGI1.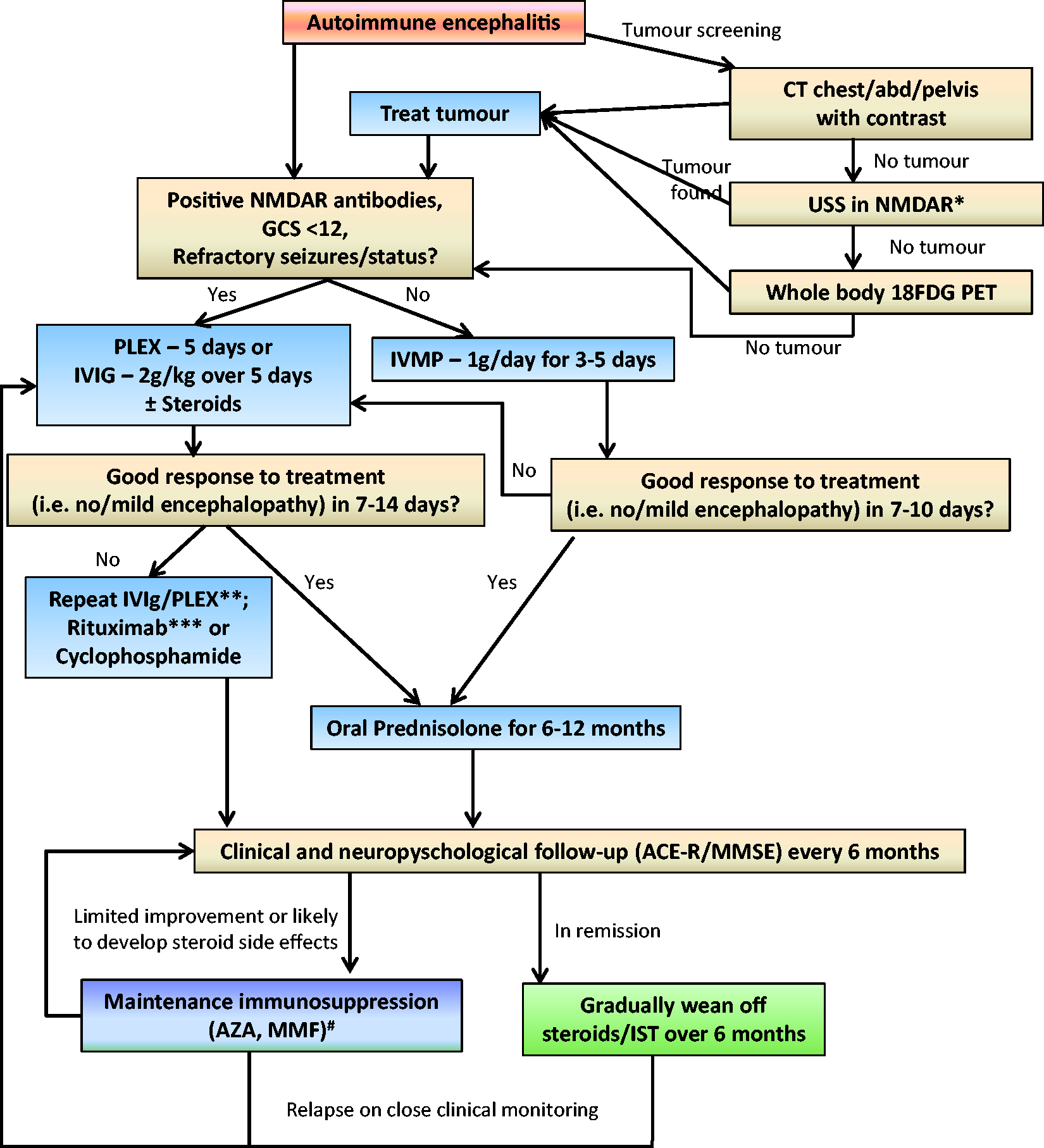
Assays
The emergence of molecular technology with higher sensitivities utilizing cell-based assays to replace the existing tissue-based methods has come at an opportune time and have been very useful for the detection of the newer markers of encephalitis discussed here.
Briefly, cDNA for various antigens are inserted into eukaryotic expression vectors (plasmid) and transfected into Human Embryonic Kidney 293 cells (HEK293). The antigen expression is only partial (between 20 and 50%), thus leaving an inbuilt negative control in cell line which is grown, fixed, validated and made commercially available by Euroimmun. This method has a rapid turnaround time. From this point onward the detection of surface (extracellular) acting autoantibodies found in serum and CSF is the same as for tissue-based assay with the exception that secondary confirmatory test is not required due to mono-specific transfection. Indirect immunofluorescence using transfected HEK293 cells has become a method of choice for the detection of cell-surface autoantibodies associated with encephalitis. Some authors suggest that the presence of CSF antibodies is more sensitive19,50 and the presence of serum antibodies alone require further confirmation using rat brain immunohistochemistry and neuronal cultures before a definite diagnosis of cell-surface antibody-mediated neurological disease is made. However, this view has not been widely supported by other authors and may not always be practically possible. Over the next few years, this should hopefully become clearer.
Our laboratory experience
In 2011, autoimmune encephalitis screening service was introduced in Birmingham. The samples are tested on Euroimmun biochip 51 with each field comprises transfected HEK293 cells expressing one of the following antigens: NMDAR, AMPAR1, AMPAR2, GABABR1, LGI1 and CASPR2. The advantage of such cell-based technology has allowed for early provision of results to the extent that in urgent cases, where the sample is in our hands we can deliver the results the same working day. This helps in quicker diagnosis and consequent accurate treatment, aiding the clinician in managing these complex neuroimmunological conditions which can mimic several other neurodegenerative and infective aetiologies.
In our hands (serum samples are diluted at 1/10 while the CSF is tested neat/undiluted), we have experienced that NMDAR transfected cells are subjected to cross-reactivity from co-presence of antinuclear antibodies (ANA) and mitochondrial antibodies (MT) which the manufacturer claim to be due to the acetone used for permeabilization and fixation. 52 However, with some experience, the NMDAR antibodies can be easily differentiated from the cross-reacting ANA and MT antibodies as the latter two antibodies will stain all the cells while the NMDAR is a cytoplasmic antibody and will only be expressed in intermittent cells. This is in contrast to the in-house methods used by some Neuroimmunology laboratories where live, unfixed and un-permeabilized transfected cells are used. The drawback of any newer test, as in this case, is the independent quality control (IQC) scheme which often lags behind due to lack of control material for distribution and laboratory demand for IQC.
Between 2011 and 2013, 452 specimens sent to our laboratory from various hospitals around the UK were tested and 5% were found positive comprising of all the above-mentioned specificities. 22 This was an unselected group of samples, and on retrospective review, some of the clinical phenotypes were not typical of autoimmune encephalitis (e.g. neuropsychiatric symptoms without typical encephalopathy, dementia and epilepsy). In the atypical cohort, even though there is a predominant neurological phenotype, it is unclear whether the antibodies are directly pathogenic or not. It emerged that among positive cases, two most common antibodies were NMDAR (43%) and LGI1 (28%). Our data confirmed published findings that these antibodies are the most common and predominantly both gender and agespecific. In this study, NMDAR was found in younger females (mean age of around 30 years) while the LGI1 was predominantly (67%) seen in males with a mean age of around 70 years. Furthermore, the clinical findings were distinct for each subtype, as has been described previously.
Conclusion/summary
Neuronal surface antibodies are recently identified important markers of various autoimmune CNS diseases. The targets of these antibodies are usually receptors or channel proteins on neuronal or glial cell surface. Clinicians should be aware of emerging neurological conditions linked to this rapidly expanding cohort of antibodies and the laboratory tests need to be done in centres with expertise to analyse the results. There still exist patients who may potentially have autoantibody-mediated neurological dysfunction and further in vitro and in vivo studies are needed to find new antigenic targets.
Footnotes
Acknowledgements
We would like to thank Dave Birch for the technical assistance. This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
None declared.
Guarantor
ARK.
Contributorship
Both authors contributed to writing the review.