
Abstract
Concerns with pubertal development are common and can cause considerable distress to patients and their carers. Many presentations reflect normal variations of pubertal timing and primarily require reassurance, although patients may opt for interventions. Other presentations need active management to avoid significant adverse effects on growth and psychosocial development. All should undergo careful assessment, particularly as some children or adolescents presenting with abnormalities in pubertal timing may have serious pathology which requires urgent investigations and treatment. This review describes the appropriate investigations and their interpretation for young people presenting with disorders in pubertal timing.
Keywords
Introduction
The assessment of disorders of puberty requires a clear understanding of the normal physiology of the hypothalamic–pituitary–gonadal (HPG) axis, and clinical changes that occur during puberty. Puberty is the sequence of physiological changes that include the development of secondary sexual characteristics associated with the pubertal growth spurt, resulting in adult stature and reproductive capacity. The onset and progression of puberty are still scored clinically according to stages defined by Marshall and Tanner.1,2 In clinical practice, the onset of puberty is defined as the presence of breast budding in girls, and testicular volumes increases to 3–4 mL in boys. Concerns with puberty usually arise when the development of physical signs are perceived to occur too early (precocious) or too late (delayed).
Precocious puberty is defined as the presence of breast development in girls or testicular enlargement of >3–4 mL in boys at 2 standard deviations (SDs) earlier than the population mean. This is accepted as before eight years in girls and before nine years in boys in the UK, but varies in different countries. The causes of precocious puberty include (Figure 1): (1) gonadotrophin-dependent precocious puberty from an early activation of the HPG axis. This is usually idiopathic in girls, but is more often pathological in boys, due in the majority to intracranial pathologies, such as brain tumours; (2) gonadotrophin-independent precocious puberty due to excess sex hormones (oestrogens or androgens) secretion from the gonads (e.g. ovarian or testicular tumour), adrenal glands (e.g. disorder of steroidogenesis pathway, adrenal tumour) or exogenous sources of sex steroids and (3) pubertal variants, which describe patients with an isolated sign of puberty without an underlying pathological disorder (e.g. isolated thelarche [breast development], premature adrenarche [pubic hair growth] and isolated menarche).
Flow diagram of investigation of precocious puberty.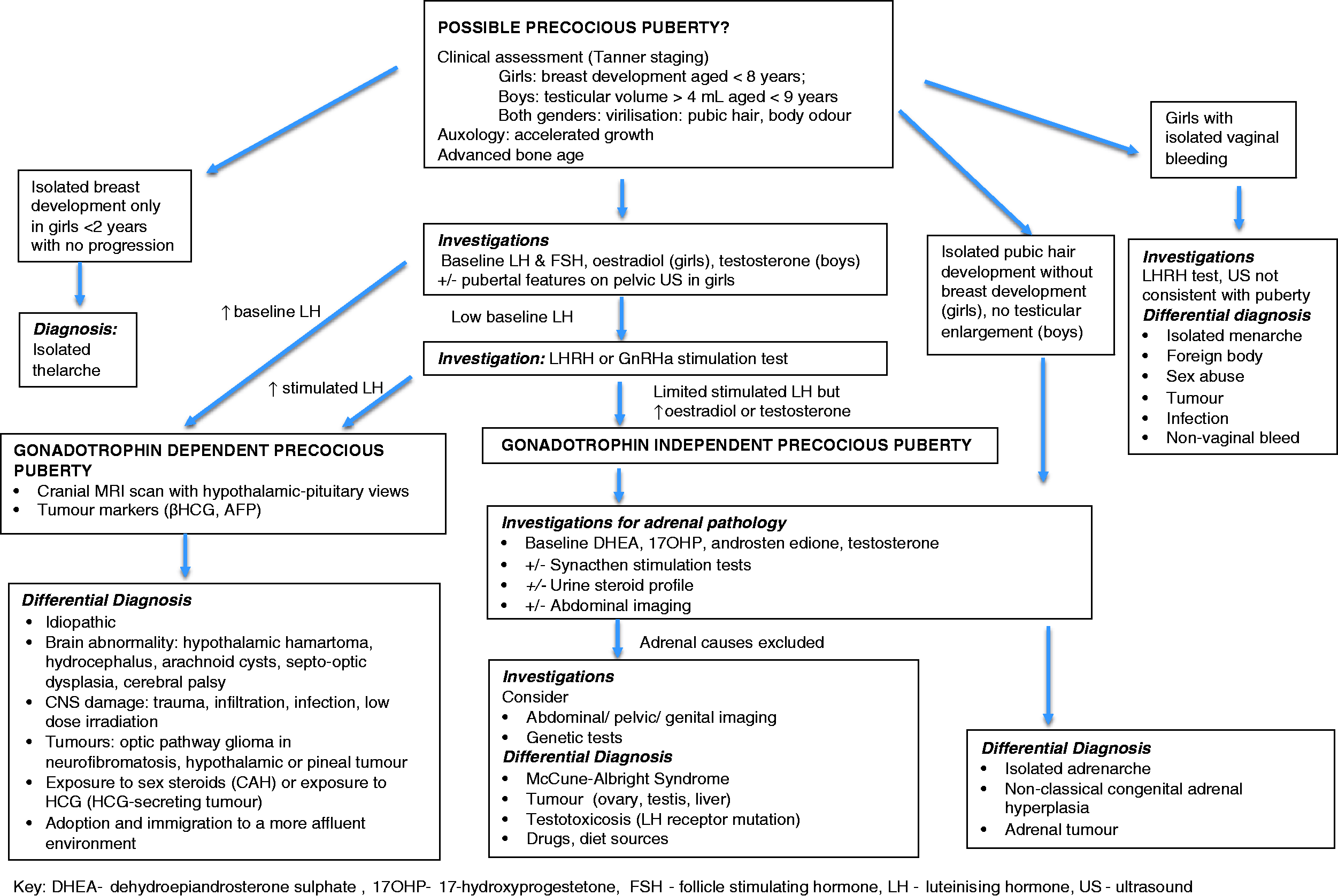
Delayed puberty is defined as the absence of testicular enlargement (<4 mL) in boys or breast budding in girls at an age 2 SD later than the population mean, which is accepted as 14 years in boys and 13 years in girls in the UK. The causes of delayed puberty include (Figure 2): (1) constitutional delay in growth and puberty (CDGP), more common in boys, is often familial due to physiological delay in HPG axis activation without associated pathology; (2) hypogonadotrophic hypogonadism due to delay or absence in central activation of the HPG axis which may be functional with associated pathology (e.g. anorexia), or hypothalamic–pituitary deficiency either congenital or secondary to pituitary damage (e.g. tumour, radiotherapy) and (3) hypergonadotrophic hypogonadism from gonadal failure or damage.
Flow diagram of investigation of delayed puberty.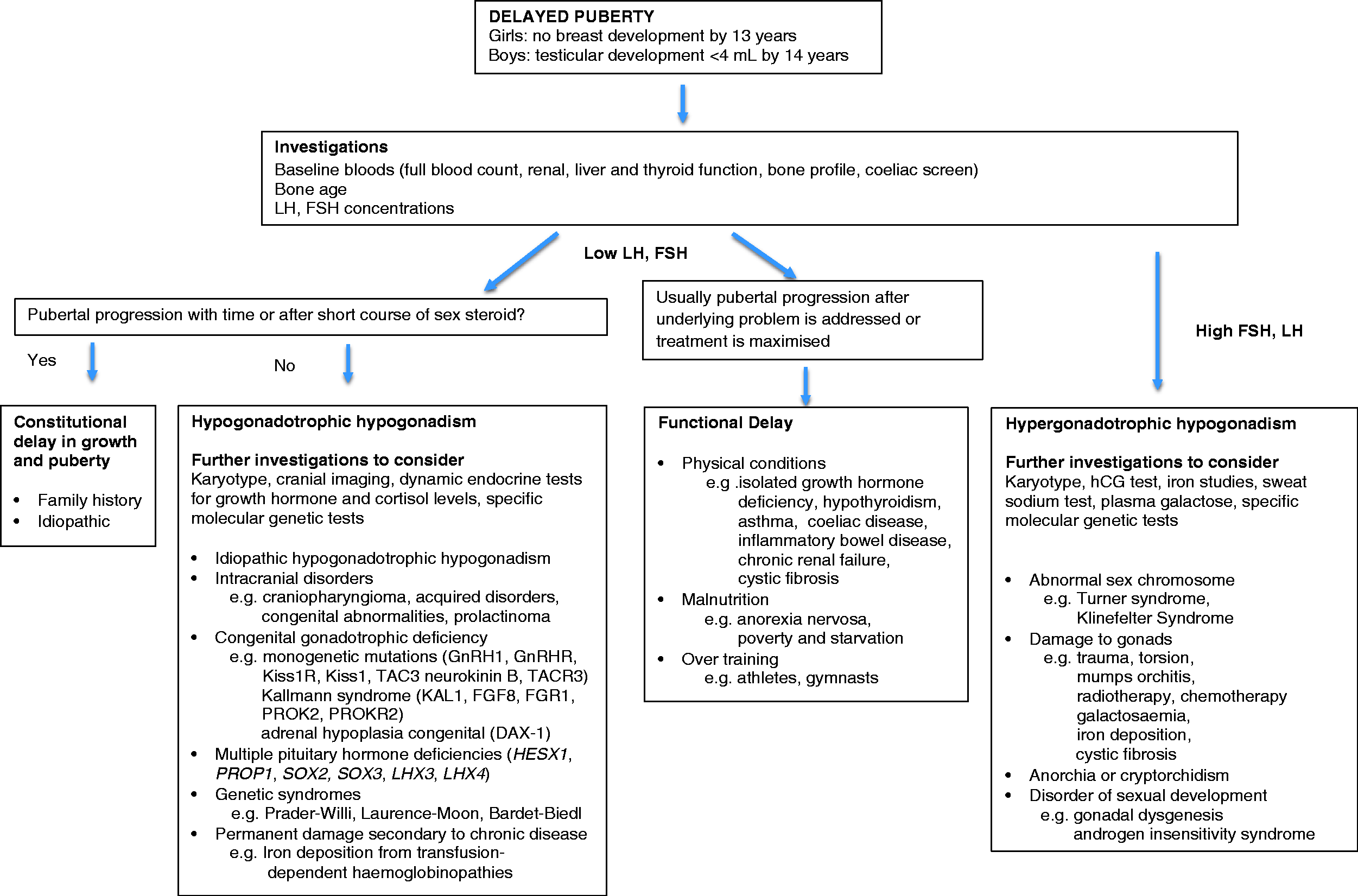
Physiology of puberty
The HPG axis is active in utero and for the first six months in boys and two years in girls, known as ‘minipuberty’. It then becomes quiescent until the onset of puberty. 3 At the onset of puberty, the hypothalamus starts to produce pulsatile secretion of gonadotrophin-releasing hormone (GnRH), which in turn stimulates the pulsatile release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. Low amplitude LH and FSH pulses initially occur during sleep and then rise with age in amplitude and frequency and extend throughout 24 h as puberty progresses.
FSH stimulates germ cell maturation, and LH produces oestrogen in girls and testosterone in boys, which lead to the development of secondary sexual characteristics that constitute puberty. In girls, puberty begins with breast development and completes with menarche. Menstrual cycles are initially irregular, but become regular over time in most females. Plasma oestradiol concentrations vary throughout the menstrual cycle. Low concentrations of oestradiol at the beginning of the cycle gradually rise to 70–260 pmol/L during the follicular phase, reaching a peak of 350–1500 pmol/L midcycle, which then drop briefly and rises again to 180–1100 pmol/L in the luteal phase. 4 In the absence of pregnancy, the corpus luteum regresses, and oestradiol concentrations drop to low concentrations at the end of the cycle as menstruation occurs. In boys, testosterone concentrations rise very slowly during childhood, but start to increase more significantly from Tanner stage 2 to an adult concentration of 10–30 nmol/L. 5 The pubertal growth spurt is the result of oestrogen-primed growth hormone production of the pituitary, with added anabolic effect of androgens in boys. This occurs earlier in girls at Tanner stage 2 compared with boys at Tanner stages 3–4.
The timing of puberty is controlled by genetic, environmental and neuroendocrine factors. Environmental factors such as nutritional status, chronic disease, migration and chemical exposure influence the regulation of puberty. Variations in the secular trend in pubertal timing have been observed in different countries. For instance, puberty continues to start earlier in some countries, but the end of puberty, especially in girls as indicated by age of menarche, has levelled off in most developed countries.6–9 A more prolonged duration of puberty (i.e. slowing of pubertal tempo) has also been demonstrated by earlier onset of breast development without changes in the median age of menarche in girls. 10 The neuroregulation of puberty is under the influence of the neuropeptides kisspeptin, neurokinin B and dynorphin A produced by neurones that are often referred together as ‘KNDy’ neurones. At the onset of puberty, the stimulatory drive of neurokinin B increases and overcomes the inhibition from dynorphin A, resulting in kisspeptin production, which is a key regulator of GnRH neurones. 11 Other neurotransmitters including gamma-aminobutyric acid (inhibitory) and glutamate (excitatory) are also implicated.
Investigations of abnormal puberty
Clinical assessment
Appropriate investigations of children and adolescents presenting with pubertal concerns must be guided by the clinical assessment including a detailed history, physical examination and review of growth. Auxology (weight, height and body mass index) must be plotted on growth charts and interpreted with reference to parental target ranges. A full physical examination must include pubertal staging and attention to general nutritional status. In boys, testicular volumes should be measured using a Prader Orchidometer. Longitudinal monitoring with serial growth and pubertal examination are important to monitor pubertal progress and height velocity.
History
In patients with precocious puberty, the clinician should enquire about features associated with androgen production (e.g. greasy skin and hair, acne, body odour, pubic hair development) and signs of sexual precocity (e.g. testicular enlargement in boys and breast development in girls). Early puberty with neurological symptoms suggests an intracranial pathology and must be promptly investigated with neuroimaging. Medical histories predisposing to precocious puberty such as neurological disorders (e.g. epilepsy, cerebral palsy), hydrocephalus, brain tumours, cranial irradiation, or conditions associated with excess endogenous androgen production, e.g. congenital adrenal hyperplasia (CAH) should be explored.
In cases of delayed puberty, the general health should be reviewed and appropriate investigations initiated to rule out the first presentation of an undiagnosed secondary pathology. Any features or medical history associated with hypogonadism such as anosmia/hyposmia, micropenis, midline defects, cryptorchidism, cleft lip/palate, bilateral orchidopexies, childhood malignancy, cystic fibrosis and transfusion-dependent conditions should be noted.
Examination and auxology
Biochemical investigations
Principles of hormone measurements
The choice of biochemical investigations should be based on the clinical picture, and good communication between the clinicians and laboratory scientists is important. Interpretation of results must take into account the laboratory methods used and the patient’s age and gender. Normative data of some immunoassays are now available for healthy infants and children.12,13 However, caution is needed in the interpretation of results in sick premature babies and also children with delayed or advanced puberty at the peripubertal ages, where age-specific reference intervals are not appropriate.
Immunoassays
At present, most hospital laboratories use immunoassays with potential limitations of reduced specificity caused by cross-reactivity with molecules of similar structures. Some immunoassays have poor sensitivity particularly at low concentrations, which can be problematic in children and adolescents as hormone concentrations are often below the limits of detection.
Mass spectrometry
Mass spectrometry is currently considered the most specific method for steroid measurement. More recent advances have involved two stages of mass spectrometry with intermediate fragmentation, often called tandem-mass spectrometry (MS/MS). Traditional methods of gas chromatography (GS) and high-performance liquid chromatography are increasingly combined with mass spectrometry. Gas chromatography-mass spectrometry (GC-MS) is commonly used to analyse metabolites of steroid hormones and their precursors from urine samples. It is non-selective and will display the entire spectrum of an individual's metabolome. However, the procedure is relatively time-consuming and expensive. Liquid chromatography tandem mass spectrometry (LC-MS/MS) measurements are targeted to single or multiple (panel) of steroids simultaneously from a small plasma volume. This is becoming more widely available for steroid hormone analysis, with reference values published for androstenedione, dehydroepiandrosterone sulphate (DHEAS), 17-hydroxyprogesterone (17-OHP) and testosterone in the paediatric age range. 5
The main advantage of LC-MS/MS is that it allows compounds of similar structures to be separated, and hence reduces the problems with cross-reactivity experienced with immunoassays. However, this has to be balanced against cost considerations.
Tests of hypothalamic–pituitary function
Basal gonadotrophins
LH and FSH are glycoproteins, each with an alpha-subunit common to LH, FSH and thyroid-stimulating hormone (TSH), and similar to human chorionic gonadotrophin (hCG) and a beta-subunit unique to each hormone.
LH and FSH are commonly measured by sandwich immunoassays which are limited by the lack of sensitivity at low concentrations, and overlapping values in prepubertal and pubertal children. Previous studies have investigated the use of a single LH measurement in the differentiation between the prepubertal and pubertal HPG axis, 14 but variations in cut-offs have been reported.
Raised LH, FSH concentrations
In patients with delayed puberty, hypergonadotrophic hypogonadism can be easily identified by raised baseline LH and FSH concentrations. Ovarian failure is usually evident by raised FSH with low or undetectable oestradiol concentrations. 15 In individuals with congenital causes, such as Turner syndrome, gonadal failure may be evident in the first one to two years of life during the minipuberty, when FSH is raised but FSH concentrations return to within the reference interval during childhood when the HPG axis is quiescent, and then rises again at a peripubertal age.
Low LH, FSH concentrations
The distinction between isolated hypogonadotrophic hypogonadism (IHH) and CDGP is more challenging due to the lack of consistent diagnostic cut-offs. 16 Some studies have reported high sensitivity and specificity using basal LH by immunoassays in the diagnosis of CDGP, but poor sensitivity especially in girls has been consistently demonstrated by others.17–19 Frequent overnight blood sampling to detect the presence or absence of LH pulses is sometimes used in research, but is impractical as a routine clinical test in children. However, FSH may be low despite the presence of primary ovarian failure, where both the gonads and hypothalamic–pituitary axis are damaged, such as, in female childhood cancer survivors treated with total body irradiation with co-existing irradiation-induced gonadotrophin deficiency.
Stimulation tests
Patients with clinical signs of precocious puberty but low concentrations of basal LH require further investigations for clarification. The luteinizing hormone-releasing hormone (LHRH) or the gonadotrophin hormone-releasing hormone analogue (GnRHa) tests are useful to identify whether the HPG axis is activated to a pubertal level. Different LHRH and GnRHa test protocols have been described.
LHRH Test
A common LHRH protocol involves measurements of FSH and LH concentrations at baseline, then 20 and 60 min after an intravenous bolus of 2.5 μg/kg or 100 μg/m2 (maximum 100 μg). 4 FSH and LH concentrations increase at 20–30 min and decrease again at 60 min. A prepubertal response includes a low LH with FSH predominance (i.e. FSH peak greater than LH). International guidelines suggested peak LH of >5 IU/L, and/or stimulated LH/FSH ratio of >0.66 as a cut-off for pubertal response during LHRH testing. 20 However, there are variations in peak LH responses depending on local assays, and lower peak LH responses have been reported in those with obesity. 21 Basal and stimulated FSH concentrations alone are not useful due to the large variability and overlapping results between prepubertal and postpubertal children. Patients with non-progressive thelarche may show some rise to LHRH in LH with a lower LH/FSH ratio than gonadotrophin-dependent precocious puberty. 20
GnRHa tests
The GnRHa test with leuprolide (500 μg s.c.) or Triptorelin (0.1 g s.c.) is used as an alternative to the LHRH test in countries, where LHRH is not available. However, large variations in the timing and concentration of peak LH which define puberty have been reported.21–23 After leuprolide administration, LH and FSH increase for 2 and 4 h, respectively, and remain high for 24 h. Peaks for testosterone and oestradiol occur later (around 16 h).
In patients with precocious puberty, a pubertal response from the LHRH or GnRHa test will be observed in gonadotrophin-dependent precocious puberty but absent in gonadotrophin-independent precocious puberty. False-positive results may be present in premature thelarche and ovarian failure.
In patients with delayed puberty, the LHRH stimulation test may not differentiate between CDGP and IHH, as there is significant overlap of LH and FSH responses between these patients. 16 Alternative protocols of the LHRH test involving administration of small repetitive pulses of LHRH over 36 h have demonstrated lower LH responses in IHH than CDGP. 16 However, these protocols are complicated and invasive with unsatisfactory diagnostic accuracy. The GnRHa test has shown better discriminatory values in differentiating IHH from CDGP, 16 which may be due to the greater stimulation of GnRHa in patients CDGP by awakening the primed normal gonadotrophic cells. However, studies are limited by small patient numbers, male gender only and lack of consistent diagnostic thresholds. 16
Tests of gonadal function
Oestradiol
Prepubertal girls have significantly higher oestradiol concentrations compared with boys, when measured using GC-MS, and this may reflect their earlier onset of puberty. 24 However, low prepubertal concentrations of oestradiol concentrations are often undetectable in immunoassays. 25 It should be borne in mind that measurements of sex steroids and gonadotrophins are not interpretable in adolescent girls taking oral contraceptive pills.
In gonadotrophin-dependent precocious puberty, oestradiol, LH and FSH concentrations rise above the prepubertal range. However, oestradiol is usually only detected at relatively advanced stages in most common assays. Raised oestradiol alone with suppressed LH, FSH response suggests gonadotrophin-independent precocious puberty, and possible differential diagnoses include exogenous source of oestrogen, a sex steroid-producing tumour, or genetic conditions associated with large ovarian cysts such as McCune-Albright syndrome.
Patients with CDGP have prepubertal concentrations of gonadotrophins and oestrogens, which will rise gradually over time. Persistently low oestradiol concentrations with raised gonadotrophin concentrations suggest primary ovarian failure, while low gonadotrophin and oestradiol concentrations suggest hypogonadotrophic hypogonadism.
Testosterone
Testosterone in the circulation is 97% bound to proteins, principally to sex hormone-binding globulin (SHBG) and weakly to albumin. Therefore, testosterone not bound to SHBG can be considered free and bioavailable. Free testosterone may be measured directly by equilibrium dialysis and ultrafiltration which are reference methods, by immunoassay, or calculated mathematically based on concentrations of total testosterone, SHBG and albumin. 26
Testosterone concentrations in prepubertal children are <1 nmol/L, which is near the detection limit of most immunoassays, but within the limits of LC-MS/MS methods of 0.1 nmol/L. 5 Only early morning samples may be useful in early puberty as testosterone production initially occurs at night, but subsequently increases throughout the day and night.27–29 The diagnosis of androgen deficiency can be challenging during puberty as the reference intervals of testosterone overlap between the different Tanner stages.5,29 Even in adulthood, testosterone concentrations fluctuate significantly between different times and days, and therefore the diagnosis must not be based on a single measurement. 29
In boys with signs of precocious puberty, raised testosterone with raised gonadotrophins indicates central causes which are more commonly pathological than in girls. The presence of testosterone excess with low gonadotrophin concentrations suggests a gonadal independent source of testosterone, such as the adrenal glands or exogenous administration or rarely an activating mutation of LH receptor. 30
Beta hCG
The hCG test assesses function of testicular tissue and defects of testosterone biosynthesis. It is sometimes used to distinguish a hypothalamic/pituitary from gonadal cause in boys with delayed puberty who have co-existing abnormalities of testicular development such as bilateral cryptorchidism, gonadal dysgenesis, anorchia (‘vanishing testis syndrome’) or other forms of disorders of sexual development.
The hCG test must be performed after the LHRH test, if they are both being done on the same day to avoid contamination of the LHRH test with hCG. The hCG stimulates testicular testosterone production from Leydig cells over a prolonged period of time via stimulation of LH receptors. A typical protocol for the short hCG test to investigate for biosynthetic disorders involves daily intramuscular injection of hCG 500–2000 IU over three days. A prolonged test over three weeks with two injections per week should be performed particularly if gonadotrophins deficiency is suspected to allow a response by receptors which may have been downregulated due to the absence of their ligand. A normal testosterone response will show a rise from baseline level but reference intervals are age and assay dependent. 4 The testosterone response is blunted, if there is poor testicular function or absent functioning testicular tissues. Elevation of androstenedione in relation to testosterone suggests a diagnosis of 17β-hydroxysteroid dehydrogenase deficiency in most 31 but not all cases. 32 Exaggerated testosterone to dihydrotestosterone (DHT) ratio is seen in 5α-reductase deficiency. 33
Inhibin B
In boys, inhibin B, secreted by Sertoli cells, demonstrates the presence of testicular tissue. Inhibin B inhibits FSH production by negative feedback. Mean inhibin B concentration in boys <1 year is 0.3 ng/L which decreases to a nadir at 6–10 years to 0.1 ng/L, and then increases to a plateau of 0.32 ng/L at 12–17 years, 34 as it becomes a marker of spermatogenesis. Low plasma concentrations are found in boys with testicular damage as a result of cryptorchidism, testicular torsion or testicular dysgenesis. 35
In girls, inhibin B concentrations reflect follicular function and the ovarian response to gonadotrophins. Inhibin B is secreted by the preantral and small antral follicles under FSH stimulation during the early follicular stage of the cycle. Inhibin B concentrations are detectable during infancy, but become undetectable in early childhood, rise again from five years, and become measurable from Tanner stage 3. After menarche, concentrations are highest at the follicular phase, and peak at ovulation. 36
In gonadotrophin-dependent precocious puberty, inhibin B concentrations are raised, but become suppressed completely during GnRHa treatment. In delayed puberty, a finding of reduced basal inhibin B concentrations has been suggested as a discriminatory test in boys to distinguish IHH from CDGP,37,38 but the study numbers were small and thresholds for the diagnosis were inconsistent.37–39
Anti-Müllerian hormone
Anti-Müllerian hormone (AMH) is a peptide homodimer measured using an enzymatically amplified two-site immunoassay. 40
In males, AMH is produced by the Sertoli cells and is responsible for fetal Müllerian duct regression and the development of the male phenotype. Median AMH concentrations are high at birth (1047 pmol/L) and increase further at 12 months of age (1082 pmol/L). They then fall gradually during childhood and declines sharply at puberty to low adult concentrations (from 697 to 49 pmol/L from Tanner stage 1 to 5). 41 In patients with 46 XY disorder of sexual development, undetectable AMH is associated with conditions caused by abnormal testicular determination (e.g. total and partial gonadal dysgenesis), but normal or elevated in impaired testosterone secretion or action. 42
In females, AMH concentrations are lower than males and are detectable from the perinatal stage to menopause. AMH is produced by developing preantral granulosa cells and small antral follicles which inhibit follicle development by negative feedback. It correlates strongly with antral follicle count, 43 and is hence a marker of ovarian follicle reserve. AMH concentrations are relatively stable throughout the menstrual cycle and are unaffected by contraception treatment. A longitudinal study of AMH concentrations measured by immunoassay in females aged 5.9 to 12.9 years showed stable concentrations maintained during childhood with median of 18 pmol/L. 44 AMH concentrations of less than 2 SDs below the mean have been shown to predict failure of the onset of puberty or imminent premature ovarian failure in Turner Syndrome. 45 AMH has also been described as a sensitive marker of ovarian function in patients with chemotherapy-induced gonadotoxicity in childhood cancer survivors. 46 In polycystic ovarian syndrome, increased AMH production leads to decreased sensitivity of FSH receptors, increased production of small 2–5 mm antral follicles, which restrain the selection of a dominant follicle, resulting in anovulatory cycles. 47
Tests of adrenal function
Blood androgens
Measurement of adrenal hormones, other than sex steroids, may be necessary in patients with signs of hyperandrogenization and possible gonadotrophin-independent precocious puberty. Adrenal causes of androgen excess include androgen-secreting tumours and enzymatic defects in the adrenal biosynthesis pathway. Some patients may present with dis-concordant development such as the presence of advanced pubic hair without testicular enlargement in boys and virilization in girls without breast development.
In addition to basal testosterone, other baseline steroid concentrations may include androstenedione, DHEAS and DHT as well as 17-OHP, and 11-deoxycortisol (11-DOC). It is now possible to measure several steroids in the same run of the LC-MS/MS so panels of steroids can be quantified simultaneously. Androstenedione is <1 nmol/L during early childhood rising in late childhood before testosterone, up to 6 nmol/L aged 13–15 years. 48 DHT follows the excursions of testosterone at a lower concentration and peaks at 3 nmol/L at Tanner stage 5. 5 Patients with equivocal results require synacthen stimulation tests and/or a urine steroid profile to confirm the underlying diagnosis.
Aldosterone and renin
To investigate for minerocorticoid activity related to a suspected adrenal disorder, plasma renin activity and aldosterone concentration are measured. Angiotensin I generation over time can be quantified by immunoassay or mass spectrometry. Some laboratories now use measurements of renin concentration. Appropriate reference intervals must be used as children have higher reference intervals than adults.49,50 Plasma renin activity or concentration is raised and aldosterone concentrations low in CAH due to a defect of 21-hydroxylase or 3β-hydroxysteroid dehydrogenase. On the contrary, in CAH due to 11-hydroxylase and 17-hydroxylase defects, high concentrations of the mineralocorticoid deoxycorticosterone lead to sodium retention and renin suppression.
Urine steroid profile
Urinary steroid profiling, usually measured by capillary GC or GC-MS, provides qualitative and quantitative data on excretion of a wide spectrum of adrenal steroid metabolites simultaneously. The test is available in specialized reference laboratories.
The highest urinary excretion rate of cortisol metabolites is between 1000 h and 1800 h. 51 There is an increase in androgen sulphate excretion and other major androgen metabolites in premature adrenarche, whereas very distinctive profiles are seen in adrenocortical tumours. 21-hydroxylase deficiency, the commonest form of CAH, shows an increase in 17-OHP and 21-deoxycortisol metabolites with low cortisol and corticosterone metabolites. 52 A urinary steroid profile enables differentiation of premature adrenarche or precocious puberty from other serious underlying pathology, i.e. inborn errors of steroid metabolism and steroid-producing tumours in patients with signs of premature virilization.
Standard synacthen stimulation test
The standard synacthen test entails stimulation of the adrenal glands by high dose (250 μg in adults) of synthetic adrenocorticotropic hormone (ACTH (1–24), also known as tetracosactide) administered either intravenously or intramuscularly, which results in a large output of cortisol and other adrenal steroids. The test is indicated in suspected adrenal insufficiency and disorders of the steroidogenesis pathway such as CAH.
The synacthen test, if performed as part of workup for abnormal puberty due to suspected steroidogenesis defects, should include interpretation of responses of cortisol and its precursors (17-OHP, 11-DOC, corticosterone, DHEA, 17-hydroxypregnenolone). A normal response is demonstrated by an adequate peak cortisol concentration of between 400 and 500 nmol/L, depending on local laboratory assays, and 17-OHP <10 nmol/L at 30 or 60 min postsynacthen administration. Patients with non-classical CAH show a 17-OHP increment of >20 nmol/L, while heterozygotes have intermediate response overlapping into the reference interval. 4 11β-hydroxylase deficiency is suggested by a rise of 11-DOC >60 nmol/L, increase of 11-DOC to cortisol ratio and decreased plasma renin activity. 53 Recent papers have reported reference intervals for plasma steroid concentrations for the rarer forms of CAH,54–56 and the overall patterns may give clues to the diagnosis57,58 ff; for example, elevation in 17-hydroxypregnenolone and DHEA in 3β-hydroxysteroid deficiency, androstenedione and androstenedione to testosterone ratio in 17β-hydroxysteroid dehydrogenase type III deficiency, and testosterone to DHT ratio in 5α-reductase type II deficiency. 59 The response to ACTH has also been shown to be capable of detecting carriers with heterozygote 21-hydroxylase gene mutations. 60
Other biochemical tests
SHBG
SHBG is a glycoprotein with high-affinity binding for 17β-hydroxysteroid hormones such as testosterone and oestradiol. SHBG is measured in girls when gonadal dysfunction may be associated with hyperandrogenism. SHBG concentrations are suppressed by insulin and androgens and stimulated by oestrogen and thyroid hormone. Reduced SHBG concentration is a surrogate marker for insulin resistance. In polycystic ovary syndrome patients with abdominal obesity, reduced SHBG concentrations result in a raised free androgen index and hyperandrogenism. 61
Prolactin
Hyperprolactinaemia in children may present as delayed puberty and in girls as secondary amenorrhoea. Hyperprolactinaemia may be caused by either disinhibition from pituitary stalk compression or excess production from a pituitary adenoma. 62 Hyperprolactinaemia inhibits GnRH secretion by increasing dopamine release from the hypothalamus and inhibits steroidogenesis. Prolactin concentrations may be raised by stress, other systemic disorders and drugs. Prolactin concentrations of more than twice the upper reference limit are usually pathological, provided the patient is not on antipsychotic medication, hypothyroid, pregnant or recently had a seizure.
Thyroid hormone
Children with undiagnosed hypothyroidism may present with growth and pubertal delay, which progress after initiation of thyroid hormone replacement. Conversely, signs of advanced puberty such as, isolated testicular enlargement in boys and isolated breast development and vaginal bleeding in girls, may be present in children with severe primary hypothyroidism at diagnosis. 63 Baseline tests classically show elevated FSH and oestradiol, but suppressed LH. The mechanism is unclear but may be caused by cross-reactivity at the structurally similar FSH and TSH receptors or direct stimulation of FSH release by elevated TSH.
Tumour markers
Tumour marker measurement is indicated when there is suspicion of an oncological process leading to precocious puberty. For example, β-hCG concentrations in suspected hCG-secreting tumour that may originate from the gonads, adrenals or liver; raised alpha-feto protein (AFP) as a marker of germ cell tumour, teratoma or hepatic tumours. 64
Non-biochemical investigations
Non-biochemical investigations for pubertal disorders are briefly discussed for completeness, but details are beyond the scope of this review.
Imaging of the central nervous system
It is important to identify intracranial lesions in patients with gonadotrophin-dependent precocious puberty. Abnormal findings on neuroimaging have been shown in 45% of girls and 92% of boys with gonadotrophin-dependent precocious puberty. 65 Cranial imaging is mandatory in the diagnostic work-up of all children with gonadotrophin-dependent precocious puberty. However, the age cut-off of routine intracranial imaging in girls with signs of precocious puberty, who are otherwise asymptomatic has been debatable.66–69 In delayed puberty, cranial imaging is also indicated if there are clinical concerns, such as headaches, growth arrest and anosmia, as this may be associated with space-occupying lesions (e.g. craniopharyngiomas) or structural abnormalities (e.g. hypoplastic olfactory bulb) of the brain.
Pelvic and abdominal ultrasound
Reference intervals of the shape and size of the uterus, ovarian volume and endometrial thicknesses on pelvic ultrasound are available to examine pelvic anatomy and maturity and signs of ovarian follicles to gonadotrophin stimulation in girls. 70 However, the parameters are not 100% sensitive and must be interpreted with the rest of the clinical and biochemical picture. 71 Pelvic and abdominal ultrasound may identify underlying adrenal abnormalities, ovarian or Leydig cell tumours 65 in patients with gonadotrophin-independent precocious puberty. In females presenting with delayed or arrested puberty, pelvic ultrasound may reveal underlying anatomical abnormalities such as absent Mullerian structures due to Mayer-Rokitansky-Küster-Hauser syndrome or 46XY complete androgen insensitivity syndrome. However, ultrasound scans of the adrenal glands are not very sensitive, and further imaging by computerized tomography or magnetic resonance imaging is recommended when there is a suspected adrenal tumour.
Bone age assessment
Bone age assessments, via X-rays of the non-dominant hand and wrist, allow comparison of skeletal maturity with chronological age, and are useful in the initial assessment, on-going monitoring and preliminary prediction of adult final height in patients with abnormal puberty. The Tanner-Whitehouse and Greulich-Pyle are the two of the commonly used methods. 72 The hand and wrist X-ray may reveal abnormal bone morphology associated with an underlying diagnosis causing delayed puberty. For example, Madelung deformity (shortened and bowed radii, long ulna leading to dorsal dislocation of distal ulna), in SHOX mutations and Turner Syndrome.
Genetic investigations
Genetic and epigenetic factors contribute to variations and abnormalities in the timing of puberty. 73 Karyotyping should be undertaken in all girls with delayed or arrested puberty to rule out Turner syndrome. Clinical signs in patients with mosaic Turner syndrome may be subtle and some may have sufficient residual ovarian function to initiate puberty spontaneously, but later present with primary amenorrhoea or premature menopause. Delayed or absent puberty in congenital hypogonadotrophic hypogonadism due to monogenic mutations (Figure 2) is rare and may be responsible in the disruption of GnRH development, migration or action. 74
Some cases of gonadotrophin-independent precocious puberty may also be associated with specific genetic mutations. For example, McCune-Albright Syndrome, a sporadic disorder caused by activating missense mutations in the α-subunit gene of the stimulatory G protein, which is characterized by the triad of gonadotrophin-independent precocious puberty, cafe-au-lait skin pigmentation and polyostotic fibrous dysplasia. 75 Testotoxicosis is a rare form of precocious puberty due to activating mutations of the LH receptor gene. 76
Diagnostic approaches to abnormal timing of puberty
This review has discussed a range of investigations that may be indicated in children and adolescents presenting with pubertal concerns. The choice of tests must be guided by the individual’s clinical picture to avoid delay in diagnosis and inappropriate use of resources. The general clinical approach and key investigations are summarized below.
Approach to patients with concerns of early puberty (Figure 1)
Patients with concerns of possible early puberty usually present with development of secondary sexual characteristics, sometimes accompanied by evidence of growth acceleration. The initial approach is to establish the diagnosis of precocious puberty and determine whether this is gonadotrophins dependent or independent, or a case of pubertal variant.
Precocious puberty
Baseline gonadotrophins and sex hormones may be sufficient to confirm the diagnosis, but this is usually not apparent until the advanced stages of puberty when the activity of the HPG axis is across day and night times. Therefore, patients with clinical signs of puberty, but low baseline gonadotrophin concentrations should undergo an LHRH or GnRHa stimulation test to confirm the diagnosis, and determine whether the sex hormone is centrally driven or not. It is then important to explore the underlying aetiology. Advancement in bone age and in girls, pubertal changes on pelvic ultrasound, may be present in precocious puberty. In gonadotrophin-dependent precocious puberty, cranial imaging and tumour markers are mandatory to rule out intracranial pathologies. Urgent referral to the neurooncology multidisciplinary team must be made, if abnormalities are detected.
Patients with gonadotrophin-independent precocious puberty require investigations to identify the source of sex steroids. Investigation of adrenal sources of sex steroids may include baseline and synacthen stimulation tests and/or urine steroid profile to exclude late onset CAH and adrenal tumours.
Tumours of the ovary, testis or liver usually presents with evidence of raised tumour markers (e.g. hCG, AFP) and will need further radiological imaging. Mutations of the LH receptor gene require genetic tests to make the diagnosis. An external source of androgen from drugs, diet or sports supplements is revealed only by detailed questioning of the child and parents.
Pubertal variants
Pubertal variants include isolated development of secondary hair (adrenarche) in both genders, or thelarche or rarely menarche in girls without other signs of puberty.
Isolated premature thelarche occurs in preschool age girls with age-appropriate linear growth without bone age advancement and absent pubic hair. Breast development may be asymmetrical, and typically waxes and wanes. Investigations are not required in mild cases. LHRH test may show a pronounced FSH peak, but LH within the prepubertal range.
Thelarche variant occurs in girls aged five to eight years and presents as persistent breast development, slight increase in height velocity and bone age advancement. Pelvic ultrasound findings may indicate oestrogen effects, but LHRH test shows prepubertal responses.
Isolated adrenarche refers to the presence of secondary hair at <8 years without breast development in girls and at <9 years without testicular enlargement in boys. It is due to the maturation of the adrenal zona reticularis, and usually occurs between six and eight years and is more common in girls who are overweight. An increase in height velocity and bone age advancement may be present. Investigations should aim to rule out abnormal sources of androgen production due to a steroidogenesis defect of the adrenal glands or adrenal tumour.
Isolated menarche is very rare. Cyclical bleeding occurs in the absence of other features of puberty. Investigations should aim to exclude precocious puberty and alternative sources of vaginal bleeding such as trauma, infection and tumour. Child sexual abuse must be considered and a referral for an examination under anaesthesia should be made when the diagnosis is unclear.
Approach to patients with concerns of delayed and arrested puberty (Figure 2)
Delayed onset of puberty
The indication for investigations of delayed puberty is strongly dependent on the clinical picture of the patient. Children with mild pubertal delay, no significant medical history and normal physical examinations are likely to have constitutional delay. There is often a family history of delayed puberty. This is a common normal variant, and extensive investigations are rarely required apart from karyotyping in all girls to exclude Turner syndrome. However, longitudinal monitoring with Tanner staging and auxological measurements are needed to ensure spontaneous onset and normal progression.
Investigations are needed in cases with more severe delay or clinical concerns. This should begin with baseline investigations, including renal, liver and thyroid functions, bone profile, coeliac disease screening, full blood count and iron studies to exclude an underlying disease. Patients with dysmorphic features and/or learning disabilities may require further genetic tests for associated syndromes such as Klinefelter, Prader-Willi, Laurence-Moon, Bardet-Biedl Syndrome (Figure 2). Endocrine tests aim to exclude abnormalities of the HPG axis as the cause of pubertal delay. In patients with gonadal failure or absent gonads, baseline gonadotrophins are raised from the peripubertal age. However, neither baseline nor stimulated gonadotrophins from LHRH or GnRHa stimulation tests can easily differentiate CDGP from hypogonadotrophic hypogonadism. CDGP may be only be distinguishable from congenital hypogonadotrophic hypogonadism via observation after the course of sex steroid treatment to see if sex steroid concentrations are maintained after treatment is discontinued in CDGP. hCG tests may be indicated in boys with under-virilization and co-existing genital abnormalities such as cryptorchidism.
Delayed puberty from hypogonadotrophic hypogonadism may be isolated or associated with an intracranial pathology. Therefore, neuroimaging with hypothalamic–pituitary views, tumour markers and prolactin concentrations are mandatory if there is clinical concern of an oncological process or a structural abnormality. As hypogonadotrophic hypogonadism may also be part of multiple pituitary hormone deficiency, patients should be assessed for other potential pituitary hormone deficiencies such as growth hormone, cortisol and thyroid deficiency and be considered for further genetic tests (Figure 2).
Pubertal arrest
Pubertal arrest is defined as the lack of pubertal progression for >2 years after spontaneous onset at the appropriate age. This may present as failure to achieve menarche from the onset of thelarche in girls, and the attainment of adult genital size (testicular volume >15 mL) from 4 mL testes in boys for >5 years. The presenting complaints may also include the lack of virilization in boys, poor breast development in girls and suboptimal pubertal growth spurt in both genders. Pubertal arrest is pathological until proven otherwise and should be assessed urgently.
Summary and conclusions
Concerns about pubertal timing in children and adolescents are common in paediatrics. Recent advances in the understanding of the mechanisms of puberty have enhanced our understanding in the pathophysiology of abnormal pubertal conditions in children. Improvements in bioassays, newer laboratory methods, biochemical markers and genetics tests have improved diagnostic yield and efficiency in many pubertal disorders. Good liaison between the laboratory scientists and clinicians is key. Clinicians must appreciate the limitations of the methodology of the tests requested and variations in local assays, and results must be interpreted with appropriate reference intervals in the clinical context of the patient. A clear understanding of the range of normal physiology of puberty and careful clinical assessments remain key to rationalize appropriate investigations to distinguish normal variants from suspected pathological conditions that require further investigations and treatment.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
ECC.
Contributorship
All authors contributed equally to the manuscript.