
Abstract
Congenital analbuminaemia is a rare autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin. The clinical diagnosis may be challenging because of the absence of unambiguous symptoms and because hypoalbuminemia may have many causes different from a genetic lack of the protein. We describe the clinical and molecular characterization of a new case of congenital analbuminaemia in an infant of apparently non-consanguineous parents from Treves, Germany. For molecular diagnosis, we used our strategy, based on the screening of the albumin gene by single-strand conformation polymorphism, heteroduplex analysis and direct DNA sequencing, which revealed that the proband is homozygous and both parents are heterozygous, for a novel G > T transversion at nucleotide c.270+ 1, the first base of intron 3. The mutation inactivates the strongly conserved GT dinucleotide at the 5′ splice site consensus sequence of this intron. In conclusion, we report the clinical findings and the molecular defect of this case, which contributes to a better understanding of the biological mechanism of congenital analbuminaemia.
Keywords
Introduction
Human serum albumin (ALB) is a single-chain, unglycosylated molecule of 585 amino acids, which comprises 60–65% of the total protein in serum, where it has a normal concentration of 35–45 g/L. 1 Its main functions are the regulation of the colloidal osmotic pressure of blood and the transport of fatty acids, hormones, metal ions, bilirubin and drugs. 1 Common causes of low circulating ALB are renal or intestinal loss (glomerular nephritis, nephrotic syndrome, protein losing gastroenteropathy), redistribution into extravascular compartments (septicaemia and other inflammatory states) and insufficient production rate (severe hepatic cirrhosis). 1 In absence of these conditions, congenital analbuminaemia (CAA, OMIM 103600) should be suspected. CAA is an autosomal recessive disorder caused by mutations within the albumin gene (ALB; GenBank accession no. NC_000004.12), which lies on chromosome 4q13.3 and is composed of 15 exons, the last of which is untranslated. 2 The clinical diagnosis of CAA is based on serum protein electrophoresis, which shows a typical pattern: minimal ALB fraction (0.001–10 g/L) and variable compensatory increases in serum globulin concentrations.3–5 The condition is characterized by surprisingly mild symptoms, such as mild oedema, hypotension, fatigue and, occasionally, a peculiar lower-body lipodystrophy, especially in adult females).3–5 Hypercholesterolemia with elevated LDL-cholesterol levels is a common finding, and some analbuminaemic individuals are treated with lipid-lowering drugs, although it is not clear whether premature atherosclerosis is present.3–5 In contrast to the benign presentation of CAA after birth, the prenatal course appears less favourable. Foetal or neonatal death of siblings was frequently noted in the families of analbuminemic individuals and CAA was shown to be a risk factor during the childhood, suggesting that ALB has a crucial role in the pre- and peri-natal period.6–7
The clinical diagnosis of CAA needs to be confirmed by mutation analysis of the ALB, which, in all so far known cases, showed that the trait is caused by homozygous or compound heterozygous inheritance of defects in the gene.4,5 Usually, heterozygous carriers, due to the presence of only one normal allele, display ALB levels near the lower limit of the normal range.3–5 CAA is very rare, since only just about 70 cases have been so far reported world-wide and are listed in the continuously updated Register of Analbuminemia Cases. 5 The frequency of the causative mutations at the heterozygous state in the normal population is higher than hitherto believed, since the most common among them, Kayseri (c228_29delAT), has an allele frequency of about six in one hundred thousand. 8
We report the clinical findings of a case of CAA in a German infant and the mutation analysis of the ALB, which revealed a novel splicing defect.
Methods
Ethical statement and clinical laboratory analyses
Subjects of this study were a German infant and his apparently non-consanguineous parents, living in a small village near Treves (Trier), located at the western border of Germany. The procedures were in accordance with the 1975 Helsinki Declaration. We collected blood samples after obtaining written informed consent from all participants involved in the study and from the parents of the proband.
ALB concentration as well as immunoglobulin concentrations were measured by nephelometry with the BN ProSpec machine (Siemens, Munich, Germany). Serum protein agarose-gel-electrophoresis was performed with the Hydrasis analyzer (Sebia, Evry Cédex, France) and measurement of total cholesterol, HDL, LDL, triglyceride as well as total protein concentrations was done by photometric tests with the AU 5400 analyzer (Beckman Coulter, Krefeld, Germany).
Mutation analysis
Fourteen genomic fragments of ALB encompassing the 14 coding exons and their intron-exon junctions were PCR amplified using specific primer pairs as described by Watkins et al.2,6 Genomic DNA from two unrelated healthy volunteers was available as a control. Heteroduplex analysis, single strand conformational polymorphism (SSCP), and DNA sequencing were performed as previously reported.9,10
Results
Clinical case
The patient is currently 2 years old. He was born after 36 6/7 gestational weeks by Caesarean section from the third pregnancy of his mother. Indications for Caesarean section were pathological cardiotocogram and suspected placental insufficiency. He was small for gestational age with a birth weight of 1.800 g, length of 45 cm and a head circumference of 30.5 cm (all measures <3rd centile).
The mother’s age is 38 years. She suffers from multiple sclerosis. The patient’s father is 43 years. They have ALB concentrations at the lower margin of the reference range, for both: 37.3 g/L. The patient has an older sister (6 years) and an older brother (4 years). They both are healthy and were not included in the present study.
Immediately after birth, the patient had thrombocytopenia leading to petechia as often seen after intrauterine growth restriction. Thrombocyte count normalized within the first days of life. However, on the eighth day of life, he started to develop subcutaneous oedema of the lower extremities, gradually worsening over the next days. Laboratory testings at that time revealed a total protein concentrations <30 g/L due to a severe hypoalbuminemia with values between 0.76 and 1.47 g/L. The patient stayed in a good clinical condition except the oedema. Extensive clinical and laboratory evaluation did neither reveal any hint for infections or inherited metabolic diseases affecting the liver’s capacities of protein production, detoxification, bile production and bile excretion nor a urinary or gastrointestinal protein loss. The patient was treated with furosemide and mother’s milk was supplemented with 0.5 g protein/dL. However, both interventions did not ameliorate the oedema. He gained weight fairly well and was sent home after 34 days. Since then he has been seen regularly as an outpatient.
Furosemide treatment and protein supplementation were stopped at the age of 3 months. The patient growth and weight gain paralleled the 3rd centile until the age of 6 months. Since then he has shown a catch up growth with a current weight and length near the 50th centile. Mental and motor development has been normal and the patient is currently attending kindergarten. The oedema regressed in the patient’s first year of life. He developed iron deficiency as he has been reluctant to eat meat. However, he has responded well to iron treatment.
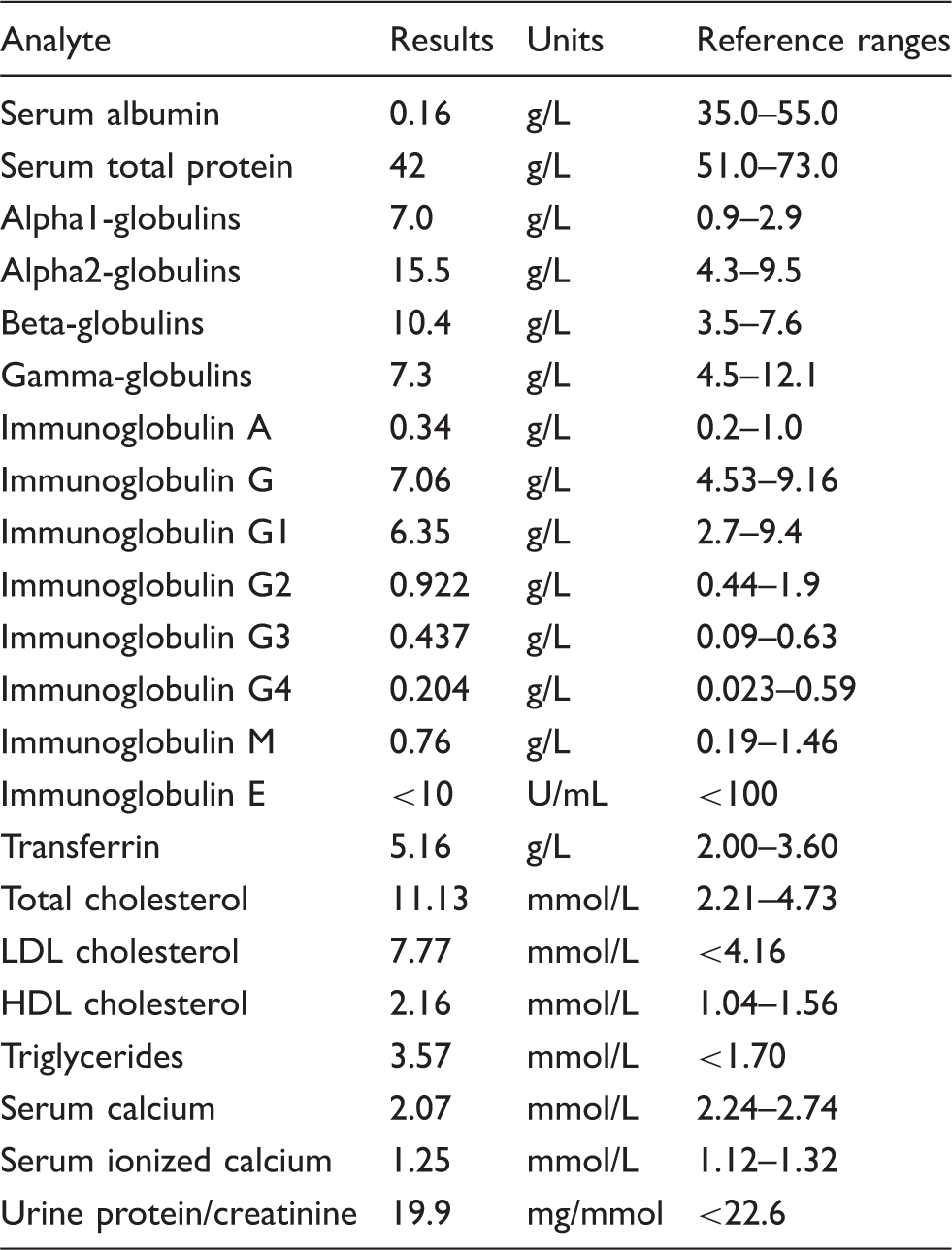
Recent relevant chemistry data of the patient are shown in Table 1. Besides the abovementioned severe hypoalbuminemia and hypoproteinaemia, the patient has extensive hyperlipidaemia, a common finding in analbuminemic subjects.
5
The immunoglobulin levels, including the IgG-subclasses are within the reference range whereas electrophoresis reveals a relative and absolute increase of the alpha 1, alpha 2 and beta-globulin fraction to compensate for the ALB deficiency (Figure 1). The patient has a total calcium concentration slightly lower than reference range, the ionized calcium concentration is within the reference ranges, suggesting that there might be binding of calcium to other up-regulated proteins.
Protein electrophoresis of the patient (a) compared to a control (b). The alpha1-, alpha2-, beta- and gamma-globulin fractions are all relatively increased. Clinical laboratory values of the analbumenic subject.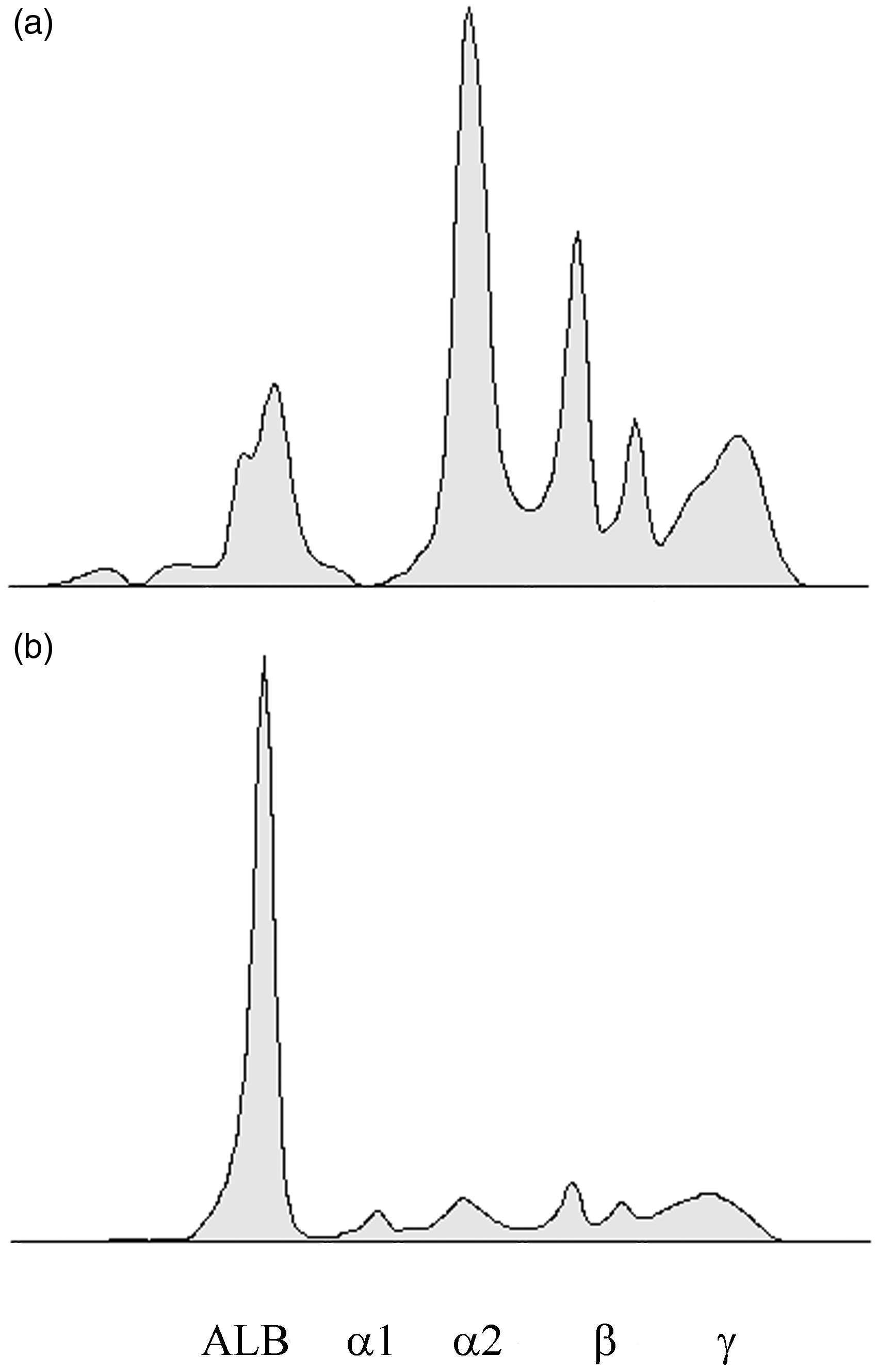
Currently, analbuminaemia does not impair the clinical condition of the presented infant with the Treves mutation. He has been put on a fat-reduced diet in order to ameliorate hyperlipidaemia. However, 3-hydroxy-3-methylglutaryl-coenzyme-A-reductase-inhibitors (statins) have not been given to him yet because his young age is a contraindication for this treatment. Besides, the benefit of a statin therapy for the patient is doubtful as statins do not reduce apolipoprotein production which is increased as a compensatory mechanism for analbuminaemia in his case. Medications with a high plasma ALB binding must be administered cautiously in order to avoid toxicity, limiting the future application of fibrates for the treatment of hyperlipidaemia, for example. Future saline infusions might disturb the patient’s current fluid equilibrium leading to severe exacerbations of oedema. 7
On the basis of the above-reported clinical and biochemical findings, CAA was diagnosed in our patient.
Mutation analysis
To confirm the diagnosis of CAA at the molecular level, a mutation analysis of ALB was carried out as described in Methods section. Both heteroduplex analysis and SSCP revealed the presence of a molecular defect in the 356 bp long region amplified by using PCR primers A05A and A06A encompassing exon 3 and the intron2–exon3 and exon3–intron3 junctions,
2
suggesting that the proband (Figure 2(a), lanes 1 and 1′) is homozygous and the parents (Figure 2(a), lanes 2 and 2′, father and lanes 3 and 3′, mother) are heterozygous for the mutation. DNA sequence analysis of this region showed that the patient is homozygous for a G > T transversion at nucleotide c.270+ 1, the first base of intron 3 (Figure 2(b)), and confirmed that both parents are heterozygous for the wild type and mutated alleles, as the sequencing electropherograms showed the presence of the two superimposed peaks at that position, representing the normal (G) and the mutated (T) base (data not shown).
Mutation analysis of the index case and of his parents. (a) Heteroduplex and SSCP analysis of exon 3. The DNA encompassing exon 3 and the exon–intron junctions from the three members of the affected family and from two controls were amplified with primers A05A and A06A and the fragments were electrophoresed onto a non-denaturing polyacrylamide gel: lane 1, proband; lane 2, father; lane 3, mother; lanes 4–5, controls. The same samples were denatured and cooled before loading: lane 1′, proband; lane 2′, mother; lane 3′, father; lanes 4′–5′, controls. (b) Genomic DNA sequence electropherograms of the analbuminemic subject. The genomic DNA was prepared as described in the Methods section. The arrow indicates the G > T transversion at nucleotide c.270+1, the first base of intron 3.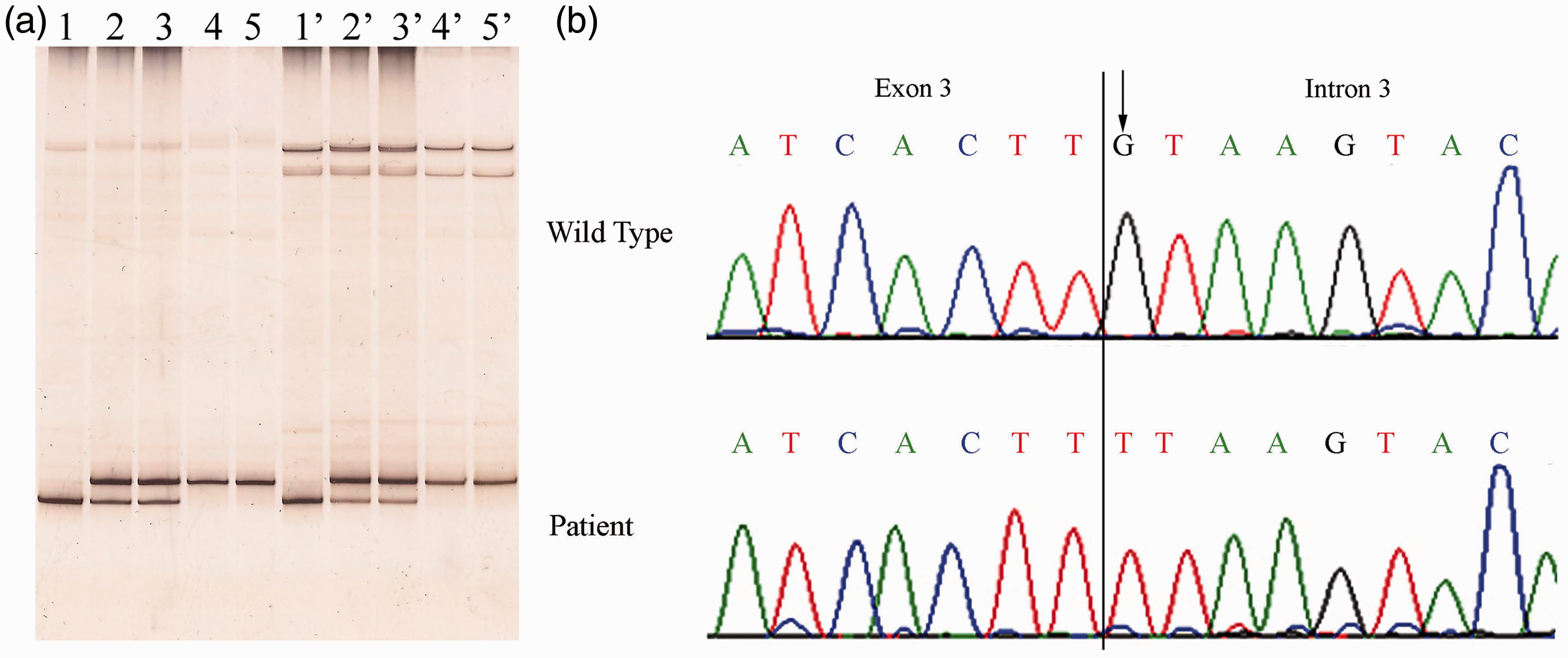
Discussion
It is well established that nearly all splice sites include invariant dinucleotides at each end of the intron, and that the 14 junctions present in the ALB conform with the GT and AG consensus sequences present at the 5′ and 3′ exon/intron splice sites, respectively. 2 The most common consequence of splicing mutations is skipping of one or more exons, followed by the activation of aberrant 5′ or 3′ splice sites and retention of full introns in mRNA. 11
For this mutation, for which we suggest the name Treves from the city of origin of the family, we could not establish the consequences of the splicing mutation at the mRNA level. This is the sixth mutation resulting in CAA, which affects the GT consensus dinucleotide sequence at the donor intron splice site, the others being Baghdad (c.79 + 1G>A), Bartin (c.1428 + 2T>C), Tripoli (c.1428 + 1G>T), Guimarães (c.1289 + 1G>A) and Ankara (c.1652 + 1G>A).4,5 In all the three cases in which the effects of the splicing mutations could be verified at the mRNA level (Bartin, Guimarães and Ankara), the result was the complete skipping of the preceding exon with a subsequent reading frame-shift within the following exon until an anticipated stop codon was found.4,5
The Treves mutation is the 23rd different molecular defect identified as a cause of CAA.4,5 Twenty-one of these were homozygous with six nonsense, eight splice site, five frame-shift/deletion, one frame-shift/insertion and one start codon mutation.4,5 The remaining two defects involved compound heterozygosity for a nonsense mutation and a splice site mutation with a subsequent reading frame-shift.4,5 Although the variant mRNA could be identified in some cases (see above), no evidence has been so far reported for the presence of the corresponding truncated protein products in the serum of analbuminaemic individuals.4,5
These 23 molecular defects are located in nine different exons and in six different introns, suggesting that the trait is the result of widely scattered random mutations. 12 However, whereas the Treves mutation, as most of the other causative defects, is so far unique, having been found in a single individual, or in members of the same family or closely related families, the two base deletion c.228_229delAT (analbuminaemia Kayseri) is by far the most frequent cause of the trait, accounting for about one third of the cases characterized at the molecular level.4,5 Therefore, the sequence c.228–c.230 of exon 3, in which also the mutation of analbuminaemia Amasya (c.229_230 delTG) is located,4,5 seems to represent the main hypermutable region in the ALB.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not required. Written informed consent from the patient was obtained.
Guarantor
LM.
Contributorship
The patient was followed by WT. Mutation analysis was performed by GC, MC, FL, MG and LM. LM and WT wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the paper.