
Abstract
Background
Systemic lupus erythematosus is a chronic multisystemic autoimmune disease characterized by chronic inflammatory processes and failure of immune-regulatory mechanisms. Systemic lupus erythematosus is associated with increased risk for cardiovascular disease. In view of immunometabolic derangements of systemic lupus erythematosus, we investigated the roles of sucrose non-fermenting AMPK related kinase, Pentraxin 3, and DNA damage in the pathogenesis of systemic lupus erythematosus complicated with cardiovascular disease.
Methods
Forty systemic lupus erythematosus women with cardiovascular disease (systemic lupus erythematosus cases), 40 systemic lupus erythematosus women without cardiovascular disease, and 40 healthy controls were enrolled in this study. Demographic and clinical data were recorded. Plasma concentrations of sucrose non-fermenting AMPK related kinase and Pentraxin 3 were immunoassayed. Carotid intima media thickness, atherogenic, and DNA damage indices were also assessed.
Results
Plasma sucrose non-fermenting AMPK related kinase and Pentraxin 3 concentrations were increased in systemic lupus erythematosus cases with cardiovascular disease compared to systemic lupus erythematosus controls and healthy controls (P < 0.0001). In systemic lupus erythematosus cases, there was a positive correlation between sucrose non-fermenting AMPK related kinase and Pentraxin 3 (r = 0.57, P < 0.002).
Conclusions
These data highlight a novel role of sucrose non-fermenting AMPK related kinase/Pentraxin 3 axis in systemic lupus erythematosus pathogenesis. Sucrose non-fermenting AMPK related kinase/Pentraxin 3 combined role in immunometabolic signaling and DNA damage response is proposed to accelerate cardiovascular complications in systemic lupus erythematosus patients.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune inflammatory disease, characterized by B/T lymphocytes, macrophages, and dendritic cells dysfunction in addition to production of autoantibodies. 1 SLE patients have elevated concentrations of pro-inflammatory cytokines such as IL-6, TNF-α and IL-1 which contribute to the inflammatoryprocess. 2 Cardiovascular disease (CVD) risk is at least doubled among SLE patients compared with the general population. 3 Atherosclerosis, a major cause of CVD, is accelerated in SLE patients due to traditional risk factors such as hypertension and dyslipidemia. 4 SLE patients possess various mechanisms to develop accelerated atherosclerosis independent of other traditional CVD risk factors, such as endothelial dysfunction, 5 immune complex generation, oxidant/anti-oxidant imbalance, and cytokine activation. 6 SLE metabolomic studies reveal evidence of heightened oxidative stress, reduced energy generation, and altered lipid profile. Persistent cellular metabolic signals promote chronic inflammation and autoimmunity. 7 The molecular basis underlying immunometabolic signaling in SLE pathogenesis remains largely elusive. 7 The Sucrose Non-Fermenting AMPK Related Kinase (SNARK) is a member of the AMP-dependent kinase family of serine/threonine kinases, which are involved in regulation of smooth muscle contractility in addition to its role as a sensor of cellular energy, and metabolism, yet its role in autoimmunity has not been clarified.8,9 SNARK RNA transcripts are induced by ultraviolet (UV) radiation in rat keratinocytes. 10 Exposure to UV radiation is among the environmental factors that have been implicated in pathogenesis of SLE. 11 Microarray data suggest that SNARK is the only kinase substantially induced in endothelial cells by TNF-α, under NF-κB transcriptional control. 12 The long pentraxin 3 (PTX3) is a member of pentraxin superfamily that plays an important role in immune system 13 and more recently is considered as a potential marker of atherosclerotic and cardiovascular disorders. 14 The proximal promoter of PTX3 contains a NF-κB binding site that is essential for its induction by TNF-α. 15 PTX3 is produced by smooth muscle cells as well as many innate immune cells such as neutrophils, fibroblasts, epithelial cells, and vascular endothelial cells. Increased concentrations of PTX3 have been detected in some autoimmune and degenerative disorders.14,16 The molecular basis of SLE pathogenesis which lead to accelerated CVD remains to be clarified. 4 This study was performed to elucidate the possible role of SNARK and PTX3 as surrogate biomarkers of atherosclerosis and CVD in Egyptian SLE patients in the context of heightened oxidative and environmental stress, altered lipid profiles, and DNA damage response.
Materials and methods
Study population
Informed written consent was obtained from all subjects enrolled in this study. This study was approved by the Research Ethical Committee of Tanta University. Eighty female SLE patients were recruited from the Department of Rheumatology & Rehabilitation, Tanta University Hospitals in the period from 2011 to 2013. Patients with SLE fulfilled the American College of Rheumatology criteria. 17 Patients were subdivided into two groups: Group I (n = 40) SLE cases who survived one or more manifestations of CVD defined as a history of myocardial infarction (non-ST-segment elevation myocardial infarction; n = 18, ST-segment elevation myocardial infarction; n = 12; five of whom underwent stenting), stable angina (n = 7), and vasculitis (n = 3). Group II (n = 40) age- and sex-matched SLE controls with no clinical manifestations of CVD. In addition, age- and sex-matched healthy volunteers within the same geographical distribution served as a healthy control group III (n = 40). All individuals were premenopausal and subjected to the same sunlight exposure and clothing conditions. Exclusion criteria included: other autoimmune diseases, tobacco smoking, oral prednisolone greater than 10 mg/day, abnormal liver or kidney function, previous transient ischemic attacks or cerebrovascular accident, and past or concurrent history of malignancy. For each subject, the SLE Disease Activity Index (SLEDAI) was assessed. 18 Assessment of SLE-related disease damage was according to the Systemic Lupus International Collaborating Clinics (SLICC) damage index. 19 None of SLE patients without CVD or apparent healthy controls were taking antihypertensive or lipid lowering drugs during the study period.
Methods
Venous blood samples were collected after an overnight fast into EDTA tubes, centrifuged at 3000 r/min for 15 min. The recovered plasma aliquots were stored at –80℃ for further analysis. All chemicals were purchased from Sigma-Aldrich, St. Louis, MO, USA, unless otherwise stated.
Routine laboratory measurements
Measurements of the following analytes were undertaken using the methods indicated: C-reactive protein (EMIT; Merck Diagnostica, Zurich, Switzerland); anti-dsDNA by ELISA kits (Immulisa, IMMCO Diagnostic, Inc., Buffalo, NY, USA); antinuclear antibody assay (Kallestad HEP-2 Kit, Biorad, USA); plasma glucose by a glucose oxidase method (Biodiagnostic, Cairo, Egypt), total lipid profile (cholesterol [TC], triglycerides [TG], and HDL-cholesterol [HDL-C] by enzymatic-colorimetric methods (Biodiagnostic, Cairo, Egypt). LDL cholesterol (LDL-C) concentration was calculated according to the Friedewald equation. 20 Atherogenic Index was calculated using the formula LDL-C/HDL-C ratio 21 and Coronary Risk Index was calculated using the formula TC/HDL-C ratio. 22
Doppler ultrasound examination
Carotid ultrasound was performed using B-mode ultrasound (Siemens G60S, Munich, Germany) equipped with a linear transducer and carotid intima media thickness (CIMT) was determined in millimeters as a surrogate measure of atherosclerosis as described. 23
Enzyme linked immunosorbent assays
Plasma PTX3 was determined using a commercial solid-phase sandwich enzyme-linked immunosorbent assay (ELISA) (Quantikine DPTX 30; R&D Systems Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions. For SNARK detection, a homemade sandwich ELISA assay was developed. Briefly, flat-bottomed ELISA plates (96 wells; Corning Glass Works, Corning, NY) were coated with 10 mg/L of anti-SNARK monoclonal antibody (sc-374348, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) diluted in coating buffer (70 mmol/L Na2CO3, 30 mmol/L NaHCO3, pH 9) overnight at 4℃. Subsequently, plates were washed three times with 0.05% Tween-phosphate buffered saline (PBST), and blocked by incubation with 3% bovine serum albumin (BSA-PBST) for 1 h at room temperature. A total of 100 µL of calibrators and undiluted plasma samples were added and incubated for 2 h at room temperature or overnight at 4℃. SNARK recombinant protein (kindly provided by M. Nakanishi) was diluted at 0, 0.0156, 0.0312, 0.0625, 0.125, 0.25, 0.5, 1 ug/L in PBST dilution buffer and served as calibrators for generation of a standard curve. After washing with PBST, 100 µL of the 1:500 v/v diluted anti-SNARK rabbit polyclonal antibody (11592-1-AP, Proteintech Group, Inc., Chicago, IL, USA) in blocking buffer was added and incubated for 2 h at room temperature. The wells were then washed and incubated with horseradish-peroxidase conjugated anti-rabbit IgG (MBL) (100 µL of 1:1000 v/v in blocking buffer for 1 h at room temperature. After washing 50 µL of HRP substrate TMB (3,3′,5,5′-tetramethylbenzidine, R&D) was added to each well and incubated for 15–30 min. Color was monitored by absorbance at 450 nm with a microplate reader (Stat Fax 2100, NY, USA).
Cell culture and immunoblotting
HeLa cells (American Type Culture Collection, Manassas, VA, USA) were cultured and maintained in Dulbecco’s modified Eagle’s medium (Gibco, Invitrogen, USA) supplemented with glutamine (2 mmol/L), 10% fetal bovine serum (Equitech-Bio, Inc., USA), penicillin (100,000 units/L; Gibco), and streptomycin (0.1 g/L; Gibco) at 37℃ in a 5% CO2 incubator. HeLa cells were either stimulated with recombinant TNF-α (20 µg/L) for 2 h, or UV (50 J/m2) (UVTEC, Cambridge), then harvested at the indicated time points. Whole cell extracts and western blotting were performed as described previously. 24 Blots were incubated with anti-SNARK antibody (11592-1-AP, ProteinTech Group, Inc., Chicago, IL, USA) at 1:100 dilution. β-actin (8226; Abcam, Cambridge, UK) was used as a loading control. Blots were washed and then incubated with respective horseradish peroxidase-conjugated secondary antibodies (1:3000) (MBL). After washings, protein expression concentrations were detected using ECL detection kit (Amersham Biosciences, Little Chalfont, UK) and chemiluminescence was detected by gel documentation system (Biometra, Goettingen, Germany).
In situ detergent extraction and immunofluorescence analysis
HeLa cells grown onto glass coverslips were treated with 400 µmol/L H2O2 at 37℃ for 3 h. Double immunofluorescence staining on paraformaldehyde-fixed cells was performed as previously described 25 using anti-phosphohistone (S139) H2AX (05-636, Millipore, Upstate, Temecula, USA) and anti-SNARK (11592-1-AP, ProteinTech Group, Inc., Chicago, IL, USA) antibodies at 1:10,000 and 1:300 dilutions, respectively. Secondary antibodies were goat anti-rabbit IgG, Alexa Fluor 488 conjugated and goat anti-rabbit IgG, Alexa Flour 594 conjugated (Molecular probes, Carlsbad, CA, USA). Nuclei were visualized with DAPI (4′,6-Diamidino-2-Phenylindole, Dilactate, Invitrogen, Carlsbad, USA). Slides were examined under fluorescent microscope (Olympus BX51, Tokyo, Japan).
DNA damage indices
Lipid peroxidation product malondialdehyde (MDA) was assayed as previously described. 26 The activity of superoxide dismutase (SOD) in plasma was assayed by commercial kit (Biodiagnostic, Cairo, Egypt). 27 For comet assay, peripheral blood mononuclear cells were isolated by Ficoll-Hypaque gradient from SLE patients or their allied controls. Alkaline comet assay was performed using a commercial kit (Trevigen’s Comet Assay kit, 4250-050-k, Gaithersburg, MD, USA) according to the manufacturer’s instructions. DNA was stained with PI and slides were digitally photographed (Olympus BX51, Tokyo, Japan).Tail moments and tail DNA contents of captured comet images were analyzed as in Park et al. 28 using TriTek (Comet Score program, Sumerduck, VA, USA).
Statistical analysis
Results are reported as mean ± SD. Multiple comparisons were performed by one-way analysis of variance followed by Tukey’s multiple comparison test. Comparison between any two groups was analyzed by the unpaired Student’s t-test or Mann–Whitney test using GraphPad Prism 5.00 software (GraphPad Software, San Diego, USA). Correlations were analyzed using Spearman rank or Pearson’s correlation coefficients. Statistical significance was considered when the P value was <0.05.
Results
Clinical and metabolic characteristics
Basic and demographic characteristics of the studied groups.
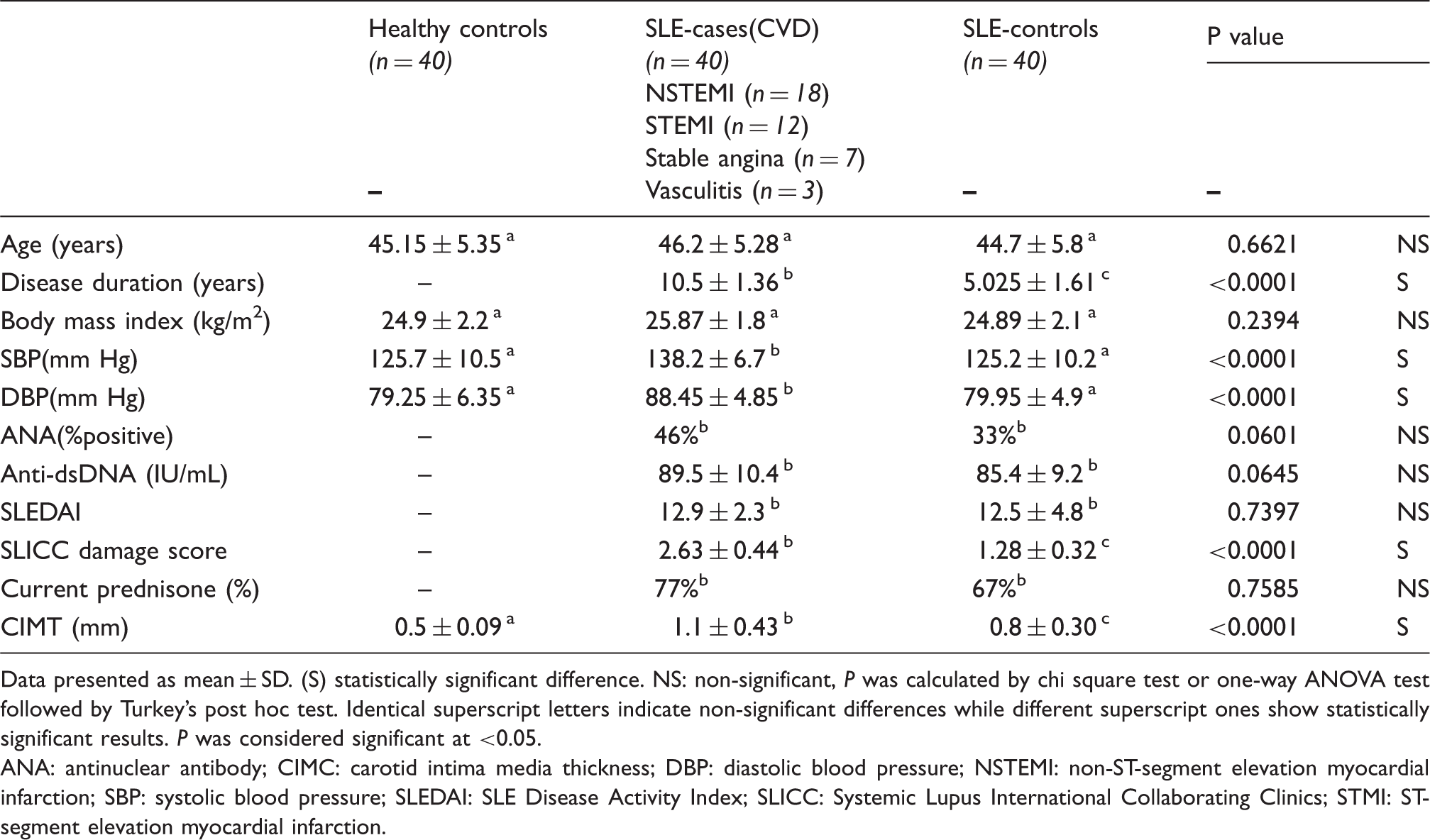
Data presented as mean ± SD. (S) statistically significant difference. NS: non-significant, P was calculated by chi square test or one-way ANOVA test followed by Tukey’s post hoc test. Identical superscript letters indicate non-significant differences while different superscript ones show statistically significant results. P was considered significant at <0.05.
ANA: antinuclear antibody; CIMC: carotid intima media thickness; DBP: diastolic blood pressure; NSTEMI: non-ST-segment elevation myocardial infarction; SBP: systolic blood pressure; SLEDAI: SLE Disease Activity Index; SLICC: Systemic Lupus International Collaborating Clinics; STMI: ST-segment elevation myocardial infarction.
Biochemical findings of the studied groups.
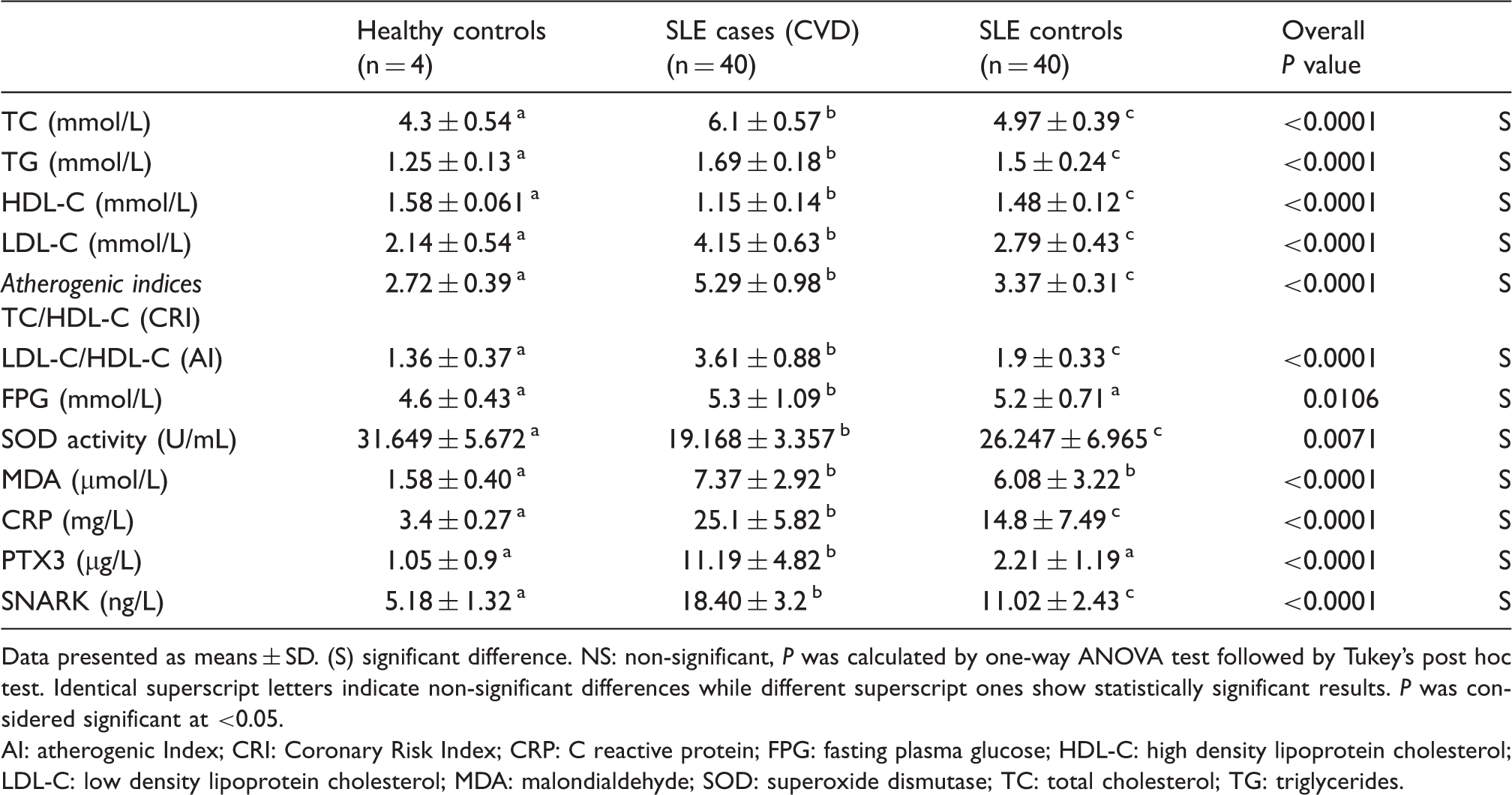
Data presented as means ± SD. (S) significant difference. NS: non-significant, P was calculated by one-way ANOVA test followed by Tukey’s post hoc test. Identical superscript letters indicate non-significant differences while different superscript ones show statistically significant results. P was considered significant at <0.05.
AI: atherogenic Index; CRI: Coronary Risk Index; CRP: C reactive protein; FPG: fasting plasma glucose; HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; MDA: malondialdehyde; SOD: superoxide dismutase; TC: total cholesterol; TG: triglycerides.
SNARK assessment
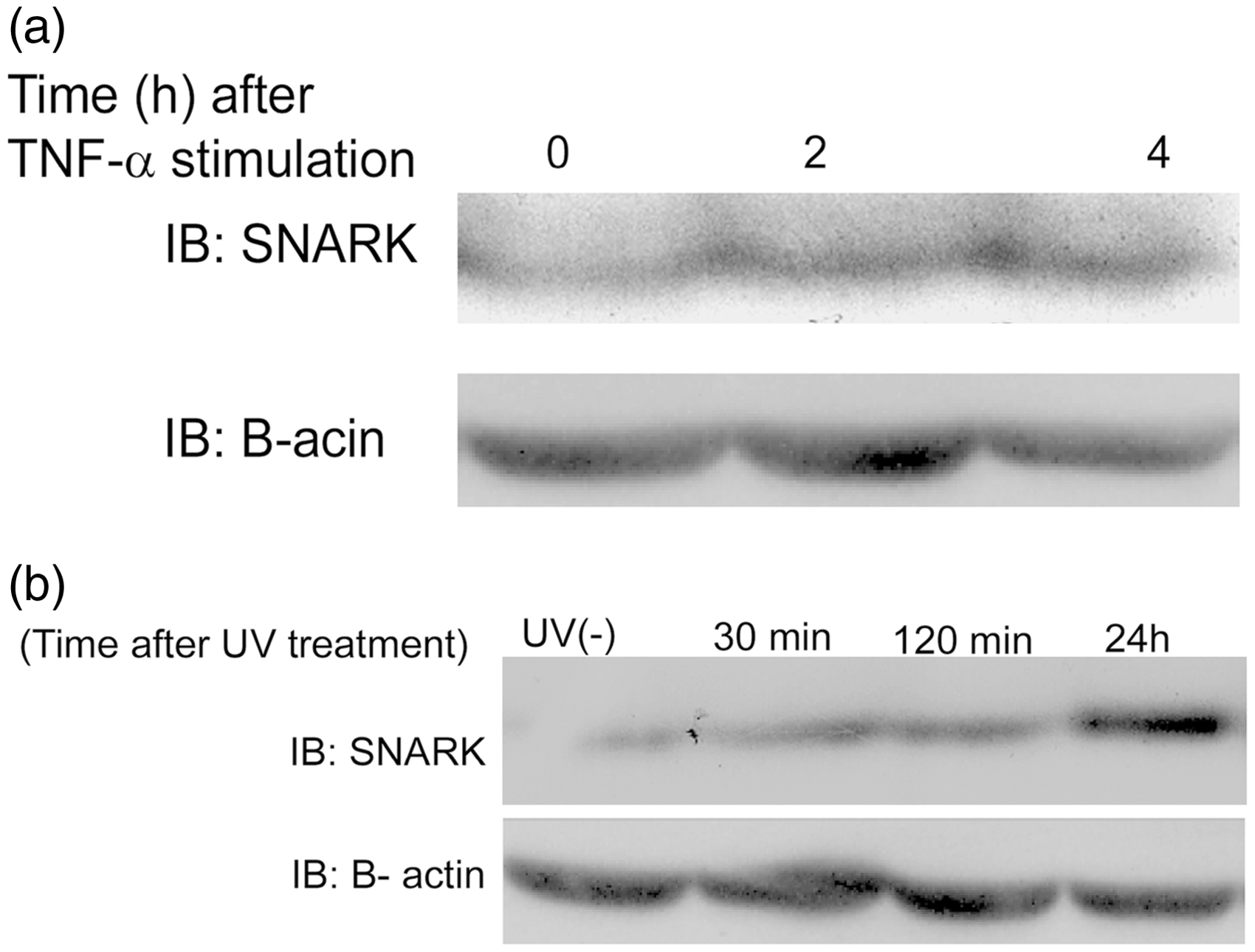
Plasma SNARK concentrations were increased in all SLE patients when compared to healthy control subjects (P < 0.0001). Furthermore, SNARK concentrations in SLE cases showed a statistically significant increase when compared to SLE controls, (P < 0.0001) (Table 2, Figure 1(a)). HeLa cells treated with TNF-α under serum starved or asynchronous conditions revealed increased SNARK expression with peak after 4 h of TNF-α treatment (Figure 4(a)).
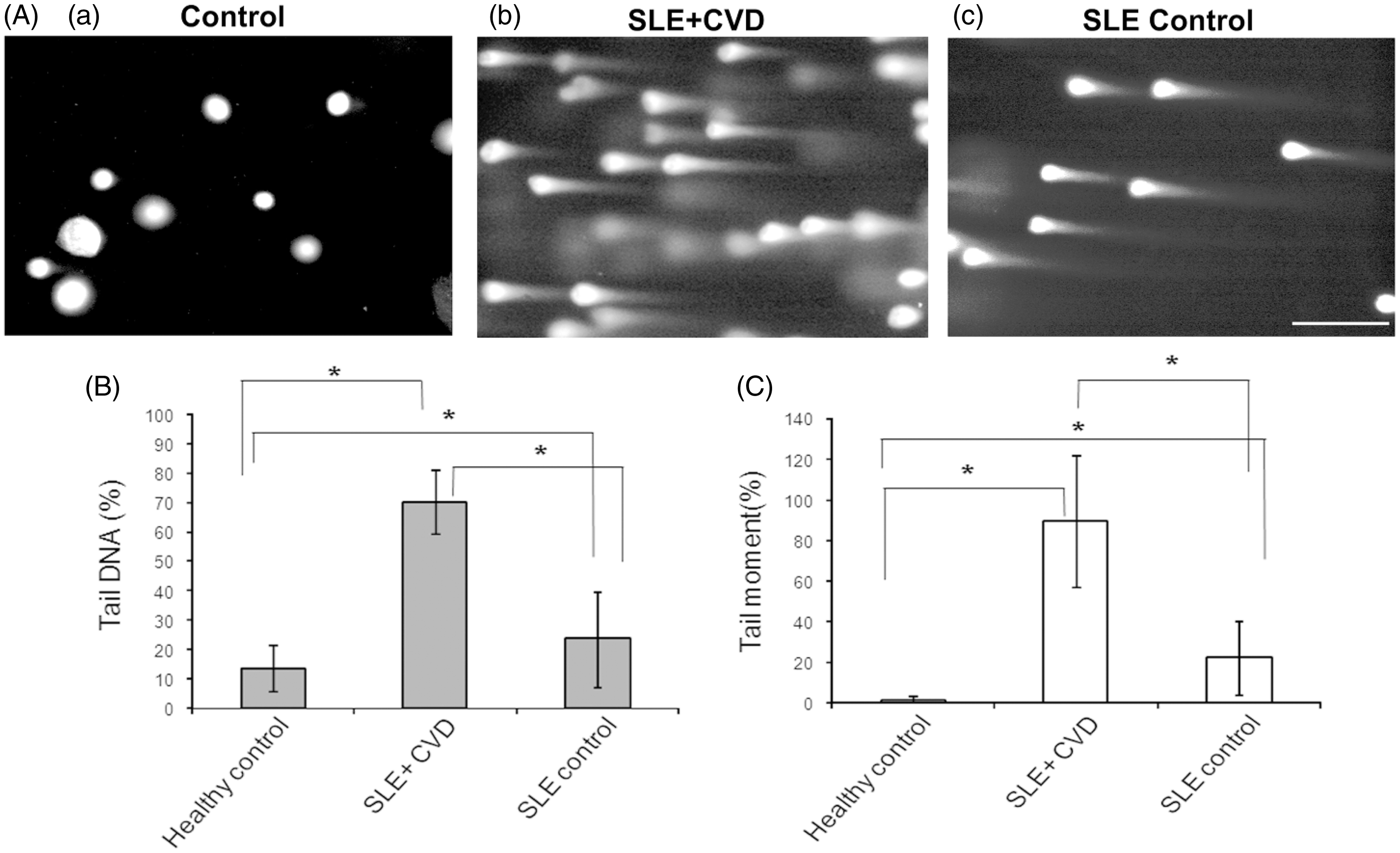
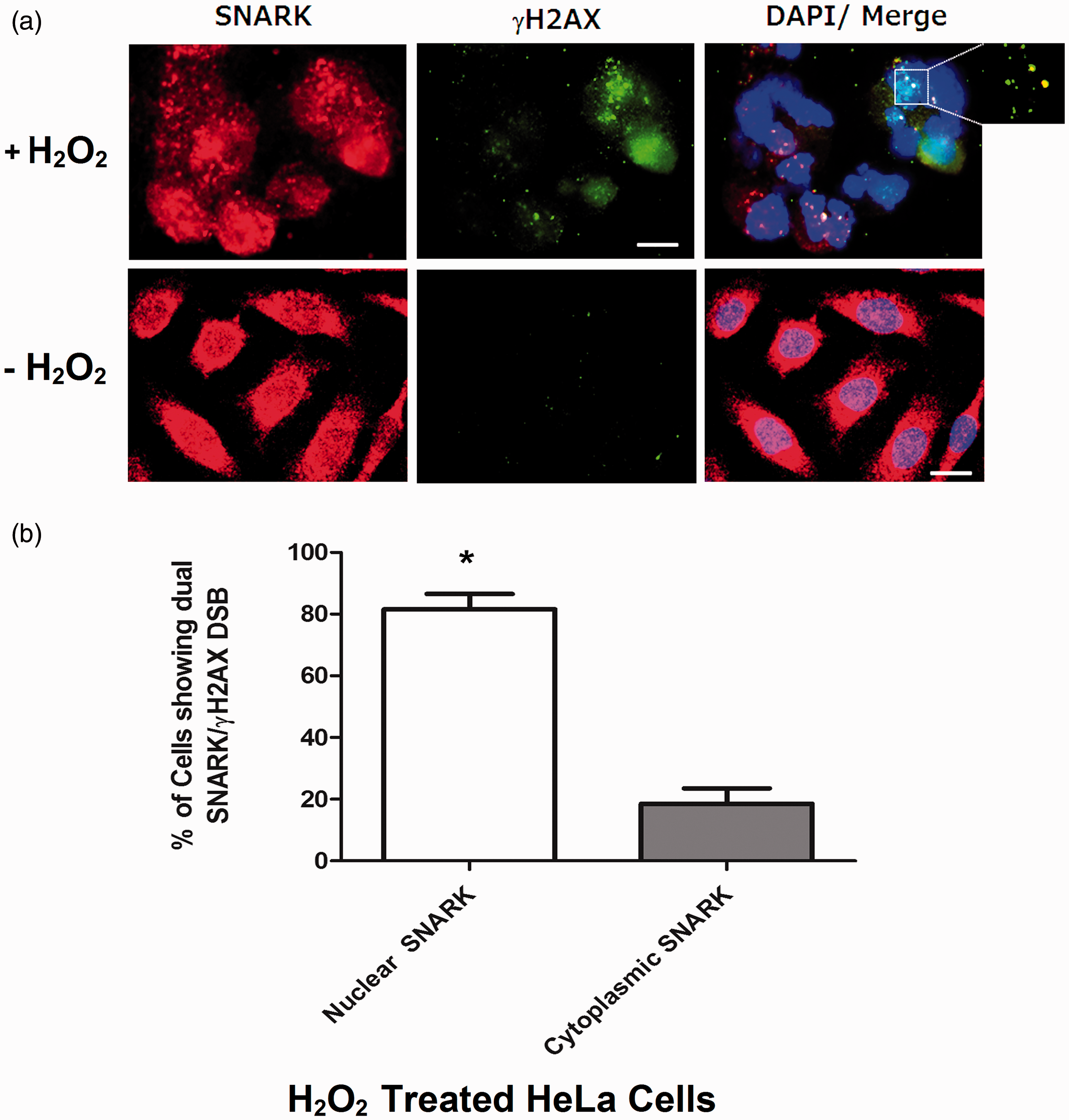
SNARK/PTX3 values in SLE patients. Graph represents mean ± SD (error bars); ***P < 0.0001 using ANOVA followed by Tukey’s post hoc test, NS: non-significant. (a) Comparison of SNARK concentrations between the indicated groups. (b) Comparison of PTX3 concentrations between the groups. (A) Photomicrographs of alkaline single cell electrophoresis (comet assay) of peripheral blood mononuclear cells from healthy controls (a), SLE cases with CVD (b), and SLE controls (c); scale bar 10 µm. (B) Tail DNA percentages in healthy control subjects, SLE cases and SLE control subjects. (C) Tail moment percentages in healthy control subjects, SLE cases and SLE control subjects. Mean ± SD, *statistically significant, P < 0.0001 by one-way ANOVA test followed by Tukey’s post hoc test. n = 50 cells scored cells from five independent experiments. The concept of tail moment = a measure of tail length X a measure of DNA in the tail as a metric for DNA migration. SNARK-mediated DNA damage response. (a) Immunofluorescence pictures of HeLa cells treated with 400 µmol/L H2O2 for 3 h (upper panel) or left untreated (lower panel), fixed and double immunostained with endogenous SNARK antibody, γH2AX antibody (DSB marker). DAPI was used to stain nuclei. Inset shows co-localization of SNARK with γH2AX at the sites of DSB. Scale bar 5 µm. (b) Graph showing percentage of cells from (a) with nuclear SNARK co-localized with γH2AX at DSBs. Data expressed as mean ± SD, (n) = 100 and results are means of three independent experiments. P value = 0.0022 by Mann–Whitney test. * Statistically significant at P < 0.005. SNARK is expressed in response to TNF-α stimulation or cellular stress. (a) HeLa cells stimulated with 20 mg/L TNF-α and cells harvested at 0, 2, 4 h post-treatment, then immunoblotted with anti-SNARK antibody. (b) HeLa cells treated with UV (50 J/m2) and harvested at 30 min, 120 min, and 24 h postirradiation, then immunoblotted with anti-SNARK antibody. (–); non-UV, β-actin was used as a loading control.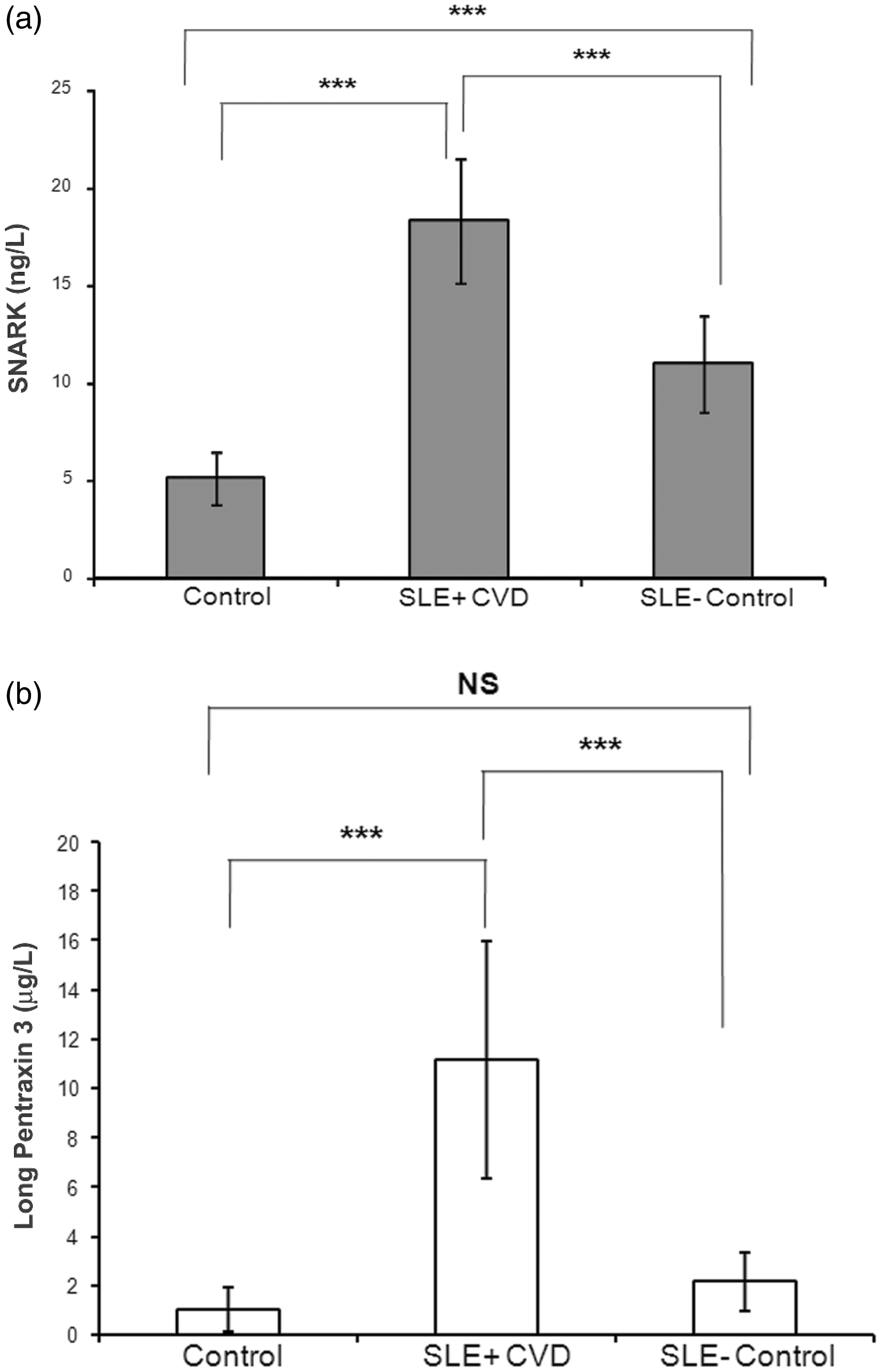
Markers of CVD
The mean CIMT was statistically higher in SLE cases as compared to SLE controls and in the latter compared to healthy subjects (Table 1). In SLE cases with CVD, plasma PTX3 concentrations were significantly higher than those of SLE controls and healthy subjects (P < 0.0001) (Figure 1(b), Table 2).
DNA damage and redox status
DNA damage in peripheral blood mononuclear cells was determined by computerized assessment of comet assay parameters and expressed as percent of tail DNA and tail moment (Figure 2(A), (a) to (c)). DNA damage was increased in cells from SLE patients (SLE cases and SLE controls) compared to healthy control subjects (Figure 2(B) and (C)). SLE cases and SLE controls had pro-oxidant/anti-oxidant imbalance represented by increased plasma concentrations of the oxidative stress biomarker MDA and a decrease in SOD activity compared to healthy control subjects. SLE cases with CVD had lower SOD activity than SLE controls (Table 2).
SNARK is involved in DNA damage response
One of the first cellular responses to double strand breaks (DSBs) is the rapid phosphorylation of the histone H2AX at serine-139 at the sites of DSBs leading to the formation of distinct gamma-H2AX foci. 29 Since SLE patients exhibited higher concentrations of SNARK as well as oxidative DNA damage, we explored the role of SNARK in DNA damage response. HeLa cells grown onto glass cover slips were treated with H2O2 or left untreated. By means of double immunostaining with SNARK and γH2AX antibodies (DSB marker), most of the human endogenous SNARK was localized mainly at the cytoplasm and interphase nuclei in untreated cells (Figure 3(a)). However, when cells were treated with H2O2, SNARK translocated into the nucleus in a foci of immunoreactivity, that co-localized with gamma H2AX foci (Figure 3(a)), and the number of cells showing positive nuclear SNARK foci co-localized with γH2AX was significantly higher than cells having cytoplasmic SNARK when cells were treated with H2O2, P = 0.0022 (Figure 3(b)), suggesting a role of SNARK in oxidative DNA damage response. In addition, we checked SNARK protein expression after UV-mediated DNA damage by means of western blotting. Increased SNARK protein expression was observed in HeLa cells treated with UV and reaches its maximum concentration 24 h post-treatment compared to untreated ones (Figure 4(b)).
Correlations of SNARK and PTX3 with some cardiovascular risk factors
Relationships of plasma SNARK concentration (µg/L) to inflammatory and cardiovascular risk factors.
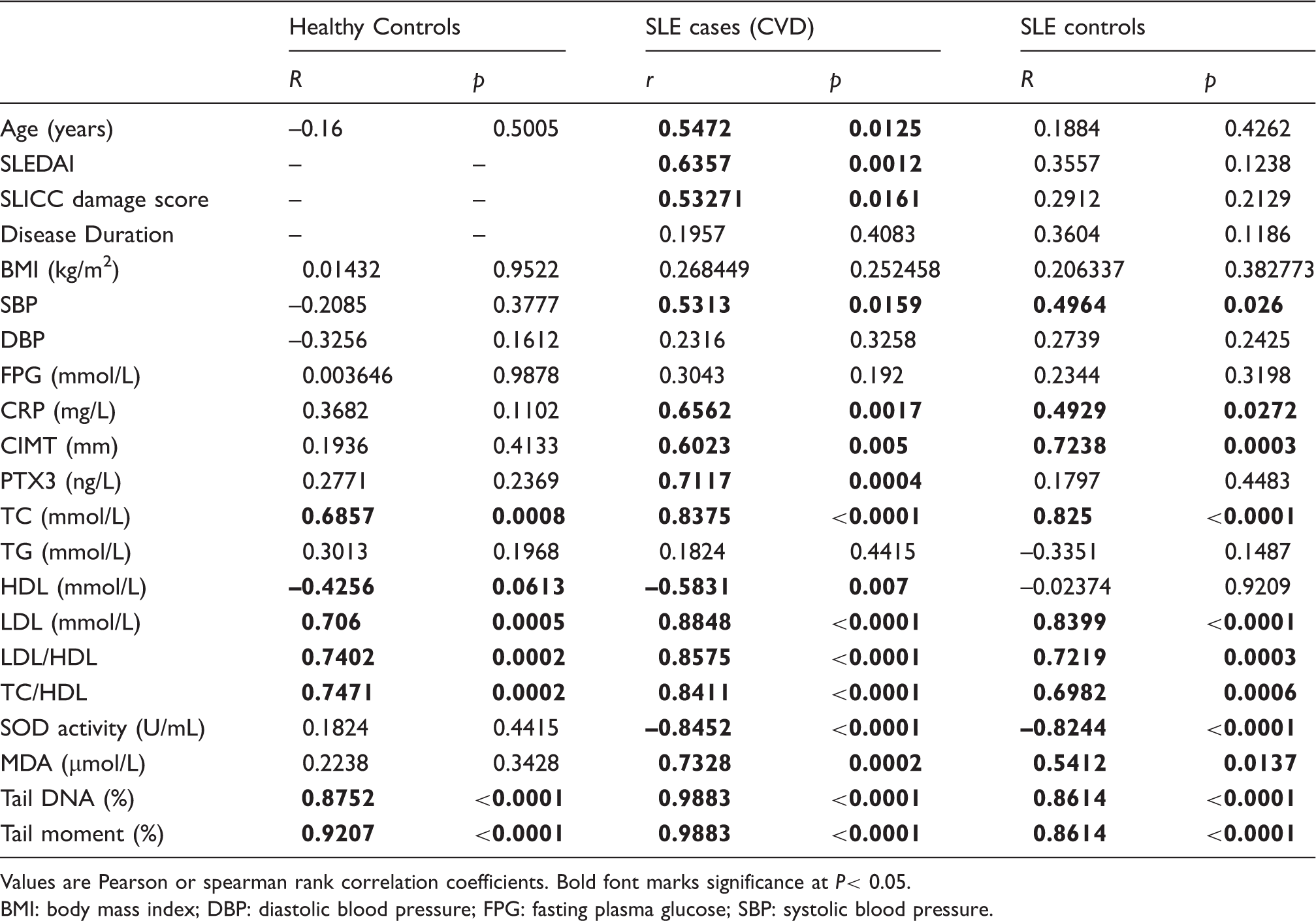
Values are Pearson or spearman rank correlation coefficients. Bold font marks significance at P< 0.05.
BMI: body mass index; DBP: diastolic blood pressure; FPG: fasting plasma glucose; SBP: systolic blood pressure.
Relationships of plasma PTX3 concentration (µg/L) to inflammatory and cardiovascular risk factors.

Values are Pearson or spearman rank correlation coefficients. Bold font marks significance at P < 0.05.
BMI: body mass index; DBP: diastolic blood pressure; SBP: systolic blood pressure.
Discussion
SLE is a chronic inflammatory autoimmune disease, where persistent cellular metabolic signals promote chronic inflammation and loss of immune tolerance.30,31 To the best of our knowledge, this study is the first to report a role for the stress/UV up-regulated kinase 10 SNARK in SLE pathogenesis. The present study clearly shows a significant increase in plasma SNARK concentrations in SLE patients relative to controls with higher values among SLE patients with CVD. SNARK expression with antiapoptotic features has been reported to be induced by CD95 via NF-κB signaling. 32 Interestingly, previous studies described increased expression of antiapoptotic genes in SLE patients in response to CD95 stimulation, 33 suggesting a role of SNARK in pathogenesis of SLE. SNARK is activated by cellular ATP depletion caused by disruption of mitochondrial or glycolytic ATP synthesis. 34 Therefore, our observed increase of SNARK concentrations in SLE patients may be due to persistent cellular metabolic signals that promote ongoing chronic inflammation and loss of immune tolerance. This is in agreement with a recent report that describes disturbances in metabolic and organelle homeostasis in SLE patients that lead to abnormal T-cell signaling and activation. 35 In our study, we observed positive correlations between SNARK and CRP, systolic blood pressure, atherogenic and DNA damage indices, as well as CIMT in SLE patients, raising a possible role for SNARK in promoting CVD complications in SLE patients. The role of SNARK in the pathogenesis of atherosclerosis remains to be clarified; however, it has been reported that SNARK mRNA expression is increased in skeletal muscle biopsies from obese humans and in cultured myotubes in response to TNF-α stimulation. 36 This is consistent with our immunoblotting results and others who detected increased SNARK expression in response to TNF-α stimulation. 37 Increased TNF-α concentrations in SLE patients has been implicated in promoting atherogenesis. 38 Moreover, increased TNF-α has been linked to coronary artery atherosclerosis with poor prognosis. 39 Intriguingly, in our cohort of SLE patients with CVD, PTX3 exhibited the highest concentrations and correlated well with SNARK concentrations, CIMT, atherogenic and DNA damage indices, as well as disease activity.
PTX3 has a role in modulation of the immunoinflammatory response associated with atherosclerosis and cardiovascular injury. Hollan et al. 40 reported that circulating PTX3 could likely be used as a biomarker for severity of CVD in inflammatory rheumatic disease. Tombetti et al. 41 reported increased PTX3 concentration in SLE and Takayasu arteritis and might represent a biomarker of actual arteritis. Consistent with our results Shimada et al. 42 reported increased PTX3 concentrations in SLE patients that was significantly associated with disease activity, however a significant correlation between PTX3 concentrations and carotid atherosclerosis was not found. This discrepancy might be attributed to different study populations and geographical distribution. In view of the ability of TNF-α to induce expression of SNARK as well as PTX3 via the transcription factor NF-κB,15,32 SNARK signaling could represent a good axis for accelerated vascular damage in SLE patients. Excessive generation of reactive oxygen species (ROS) in SLE has the potential to initiate modification and damage to lipids, proteins, and DNA with generation of auto-antigens which can elicit autoimmune responses leading to chronic inflammation and CVD. 43 Lipid peroxidation generates a variety of relatively stable end products such as MDA that induce DNA damage. 44 In this study, increased MDA concentrations were noted in cases with CVD, a finding that is in accordance with that obtained previously. 45 Furthermore, the present study shows evidence of reduction in SOD activity in SLE cases with CVD compared to other groups. This could be due to the presence of autoantibodies against SOD which lead to the formation of immune complexes. 46 Additionally, we detect higher concentrations of DNA damage among SLE cases. In support of our findings, prior studies have revealed an abnormal DNA damage response in autoimmune diseases including SLE. 47 Our observed positive correlation between SNARK and DNA damage indices drives us to characterize a possible role for SNARK in DNA damage response. We demonstrated that SNARK is translocated to the nucleus in response to oxidative DNA damage and is recruited to chromatin at sites of DSB, predicting a role of SNARK in oxidative DNA damage response and strengthening the notion that SNARK may act as a modulator of gene expression through its nuclear localization and chromatin binding. 48 Environmental stresses, such as UV have been implicated in SLE pathogenesis, 49 by inhibiting DNA methylation that convert normal antigen-specific CD4+T lymphocytes into autoreactive, cytotoxic, pro-inflammatory cells and consequently accelerates lupus development and activity. 49 This is consistent with our observation, where SNARK protein expression is increased in response to UV treatment. This could be mediated via NF-κB activation that in turn increased SNARK transcription 32 and mechanistically unravel a new role for SNARK in SLE pathogenesis. Importantly, our study demonstrates altered lipid profiles and increased atherogenic ratios among all SLE patients studied. These changes are associated with increased incidence of CVD in the general population. 50 Previous work stated that endothelial dysfunction in SLE patients was associated with abnormal lipid profiles. 23
In SLE patients with CVD, SNARK concentrations correlated positively with CIMT and LDL concentrations that explain its possible role in promoting early atherogenesis and cardiovascular complications in SLE patients. Surprisingly, our SLE cases had higher CIMT values than SLE controls, who had been on long-term steroid therapy indicating that steroid therapy alone may not be associated with premature CVD in SLE, a finding that has been previously reported 51 and allows the possibility that certain signaling pathway(s) might initiate atherogenesis and CVD risk in certain subsets of lupus patients. Rho-associated protein kinase (ROCK) has been implicated in autoimmunity and SLE pathogenesis. 52 ROCK mediates its actions partially through phosphorylation of the smooth muscle regulatory subunit myosin phosphatase target protein-1 (MYPT1) that promotes endothelial cell contraction, permeability, and atherosclerosis. 53 Interestingly, SNARK also phosphorylates MYPT1 on other sites, 37 and regulates actin stress fibers formation, 9 highlighting an atherogenic potential of SNARK mediated by modulation of endothelial function and smooth muscle contractility. A schematic model, depicting our proposed SNARK/PTX3 signaling is reported in Supplementary Figure 1, where we suggest that varying signals such as ATP depletion, TNF-α, UV light, and ROS might result in increased SNARK expression. SNARK involvement in SLE pathogenesis may affect B-cell homeostasis, control smooth muscle contractility, or control gene expression of some target(s). The synergetic increase of SNARK/PTX3 via TNF-α/NF-κB signaling and DNA damage response may mediate SLE pathogenesis, atherogenesis, and CVD development. The novel proposed role of SNARK/PTX3 opens the door for further diagnostic and therapeutic approaches that ensure modification of metabolic and immune-modulatory status to prevent premature cardiovascular complications in SLE patients.
Footnotes
Acknowledgements
We would like to thank Dr Safwat M and Prof Nakanishi M (Nagoya City University, Japan) for providing fluorescent antibodies and human recombinant Nuak2 protein, Dr Miura Y, (Nagoya City University, Japan) for help in providing chemicals and reagents, Dr Zaytoun H, (Radiology Department, Tanta University) for Doppler ultrasound examination.
Conflict of interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
The Ethical Committee of Tanta Faculty of Medicine approved this study (2868/11/14).
Guarantor
DHZ.
Contributorship
DHZ researched literature and conceived the study, performed experimental work, and wrote the first draft paper. AME-B was involved in protocol development, gaining ethical approval, patient recruitment, and data analysis. WAK contributed to experimental work, statistical analysis, and to the writing of the manuscript. AHG helped in clinical cardiovascular assessment of data. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.