
Abstract
Background
Alpha 1 antitrypsin (A1AT) is the major plasma serine protease inhibitor that is produced in liver cells. A1AT deficiency is recognized globally as a common genetic cause of liver disease in children, which results from mutations in the SERine Protease INhibitor A1 (SERPINA1) gene. The importance of A1AT deficiency in Viet Nam is unclear. The aim of this study was to determine the A1AT variants present in paediatric patients with liver diseases in order to clarify whether A1AT deficiency is present in Viet Nam.
Methods
A1AT studies were carried out in 130 children with liver disease of indeterminate aetiology. A1AT levels were determined by immunoturbidimetry. Phenotype analysis of A1AT was performed by isoelectric focusing (IEF) in all patients. Genotype analyses to determine A1AT mutations were performed by direct sequencing.
Results
We identified a rare variant of A1AT named Zbristol. The Zbristol appeared to be deficient in the plasma to about the same degree as the PI S protein resulting in low concentration of A1AT in one of these two Vietnamese patients. No other deficient A1AT allele was detected, although 11 patients (8.5%) showed a reduced serum concentration of A1AT.
Conclusions
These are the first two cases of a rare A1AT deficiency allele to be found in Viet Nam clearly inferring that A1AT deficiency is not just a disease of Caucasians. As such, the laboratory diagnosis of A1AT deficiency including A1AT concentration determination and phenotype and genotype testing should form part of the routine differential diagnosis of paediatric liver disease of indeterminate aetiology in Vietnamese patients.
Introduction
Alpha 1 antitrypsin (A1AT), also known as alpha 1 proteinase inhibitor (α1-PI), is a glycoprotein which functions as a serine protease inhibitor (PI). A1AT is synthesized primarily by the liver and then released into the bloodstream. A1AT’s major physiological target is neutrophil elastase (NE), a protease that is actively secreted from neutrophils during periods of inflammation. NE degrades the elastin of alveolar walls and other structural proteins in a variety of tissues. A1AT’s primary action is therefore protective as it blocks the effects of NE on these surfaces. 1 A1AT is encoded by SERine Protease INhibitor A1 (SERPINA1) gene (14q32.1). It spans 12.2 kb and is organized into four coding exons (2, 3, 4 and 5) and three noncoding exons (1a, 1b and 1c). It is a highly polymorphic gene with more than 100 variants reported. A1AT variants are co-dominantly inherited. The PI M allele is the wild type and is the most prevalent. A proportion of these variants have an effect on A1AT concentration or function. Such gene mutations can result in the production of an abnormal form of the A1AT protein that cannot be released from the liver, resulting in low levels in the blood. 1,2 The clinical disease associated with A1AT deficiency can present in a number of ways, but the most frequent organs affected are the lung and the liver. In the lung, emphysema is the most common manifestation. However, the spectrum of liver diseases associated with A1AT deficiency includes neonatal cholestasis, chronic hepatitis and cirrhosis in children and hepatocellular carcinoma later in life. The PI Z allele (Gly342Lys) is the most common variant associated with deficiency in Caucasians. 1,3 However, although A1AT deficiency is among the most common hereditary disorders on a global scale, the importance of A1AT deficiency in Viet Nam is unclear. In fact, many Vietnamese children with liver disease are of indeterminate aetiology. The aim of this study was to determine the A1AT variants present in paediatric patients with liver diseases (neonatal cholestasis and other chronic liver diseases) in order to clarify whether A1AT deficiency is present in Viet Nam.
Materials and methods
Study group
A total of 130 venous blood samples were collected from paediatric liver disease patients who were hospitalized in the Department of Gastroenterology and Hepatology of the National Hospital of Pediatrics and Saint Paul General Hospital during the period August 2008 to July 2011. Blood samples from a control population of 30 paediatric patients were included for comparative study. These subjects (same ages as patients) were hospitalized for causes other than liver diseases. Informed consent was obtained from all patients’ guardians.
A1AT quantification, phenotyping and genotyping
Blood samples were collected when patients had no symptoms of acute inflammation to minimize any increase in A1AT level due to an acute phase response. The serum samples were separated by centrifugation and stored at −80℃ until analysis. A1AT concentrations were determined by immunoturbidimetry on an Olympus AU400 analyser (Beckman Coulter, Inc., distributed by Mitalab Company Limited, Ha Noi, Viet Nam). The testing procedure was carefully monitored and quality control of the test was done in each run, following the manufacturer’s instructions. The assay’s imprecision was 2.2% and 2.0% at 0.40 g/L and 2.98 g/L, respectively. The assay is able to determine A1AT concentrations between 0.30 g/L and 5.0 g/L, with its accuracy traceable to Certified Reference Material 470 calibrator. The literature recommended reference interval of 0.90–2.0 g/L was applied, with the lower limit of 0.9 g/L used as the cut-off value to define A1AT deficiency. 4,5 We have also analysed A1AT phenotypes for all of the patients and controls by Isoelectric Focusing (IEF) on IsoGel 1% with pH gradient 4.2–4.9 on MINI IEF CELL model 111 (Bio-Rad Laboratories, Inc. distributed by DKSH Vietnam Co. Ltd, Ho Chi Minh City, Viet Nam). The A1AT bands were detected by immunofixation and staining with Coomassie™ Blue PhastGel™ R-350 (GE Healthcare Life Sciences, distributed by Medi Group Asia Ltd, Ha Noi, Viet Nam). The stained band patterns were visually examined to determine the A1AT protein phenotype in comparison with control samples of known A1AT phenotypes which were kindly provided by Monash Medical Centre (Australia). We next screened by sequencing the coding region of SERPINA1 with exons 2 and 5 on 38 patients, i.e. 11 with serum level of A1AT below the cut-off of 0.90 g/L, six with phenotypes other than PI MM and 21 with normal A1AT serum levels (randomly selected). The DNA extraction from whole blood samples was performed using the QIAamp DNA blood mini kit (QIAGEN, Germany distributed by Viet Anh Instruments Co., Ltd, Ha Noi, Viet Nam), according to the manufacturer’s instructions. Relevant parts of the SERPINA1 gene (exon 2 and exon 5) were amplified and sequenced using the following primers: 2 F: 5′-ATG CTG CCC AGA AGA CAG-3′; 2 R: 5′-CTA TGG GAA ACA GCT CAG G-3′; 5 F: 5′-AGC CTT ACA ACG TGT CTC TGC-3′; 5 R: 5′-GGA TTT ACA GAT CAC ATG CAG G-3′. The sequences of primers were obtained from previously published sequences. 6 Polymerase chain reaction (PCR) conditions reaction mixtures contained 10 × buffer, 2.5 mM of dNTP, 10 pmol of each primer and 5 U/µL of Taq polymerase (VNTAB Co., Ltd Ha Noi Viet Nam). The thermocycling program included a 3-min incubation at 94℃, followed by 30 cycles of 94℃ for 45 s, 55℃ for 45 s and 72℃ for 45 s, finished by 72℃ for 8 min. Purified PCR products were sequenced with an Applied Biosystems 3730XL DNA Analyser (Macrogen Inc., Seoul, Korea).
Data analysis
Sensitivity and specificity calculations were made using the on line program from Vassar (http://faculty.vassar.edu/lowry/clin1.html). The Microsoft Office Excel software (FPT Distribution Co. Ltd, Ha Noi, Viet Nam) was used for statistical analysis of the serum A1AT concentrations in the liver diseases group and control population.
Results
We investigated the correlation between serum A1AT concentration and phenotypes in all patients and the control subjects. The age of the 130 enrolled patients ranged from two weeks to 24 months (70% patients under three months), and ratio of male to female babies was 1.95, including 86 boy babies (66.1%) and 44 girl babies (33.9%).
Immunoturbidimetric assay revealed that the serum A1AT concentration of the control subjects ranged from 0.93 to 2.22 g/L (median 1.48 g/L) whilst the serum A1AT concentration of the patients ranged from 0.40 to 2.12 g/L (median 1.32 g/L). Eleven (8.5%) patients showed a reduced serum concentration of A1AT below the cut-off of 0.90 g/L. Of these 11 infants, seven patients were just below the cut off; two patients had moderately reduced levels between 0.45 g/L and 0.80 g/L; and two patients were severely deficient with serum A1AT levels <0.45 g/L (Figure 1).
Serum A1AT levels determined in 130 liver disease patients by immunoturbidimetric assay. Each dot reveals the A1AT concentration of each patient, with the PI MM (normal variant), PI MX Christchurch (rare normal variant) and PI MZbristol index patients identified separately. The serum A1AT levels of the patients ranged from 0.40 to 2.12 g/L, median 1.32 g/L. An A1AT level of <0.90 g/L (circled) was considered deficient. The serum A1AT levels of the 30 control subjects with PI MM phenotype ranged from 0.93 to 2.22 g/L, median 1.48 g/L (data not shown).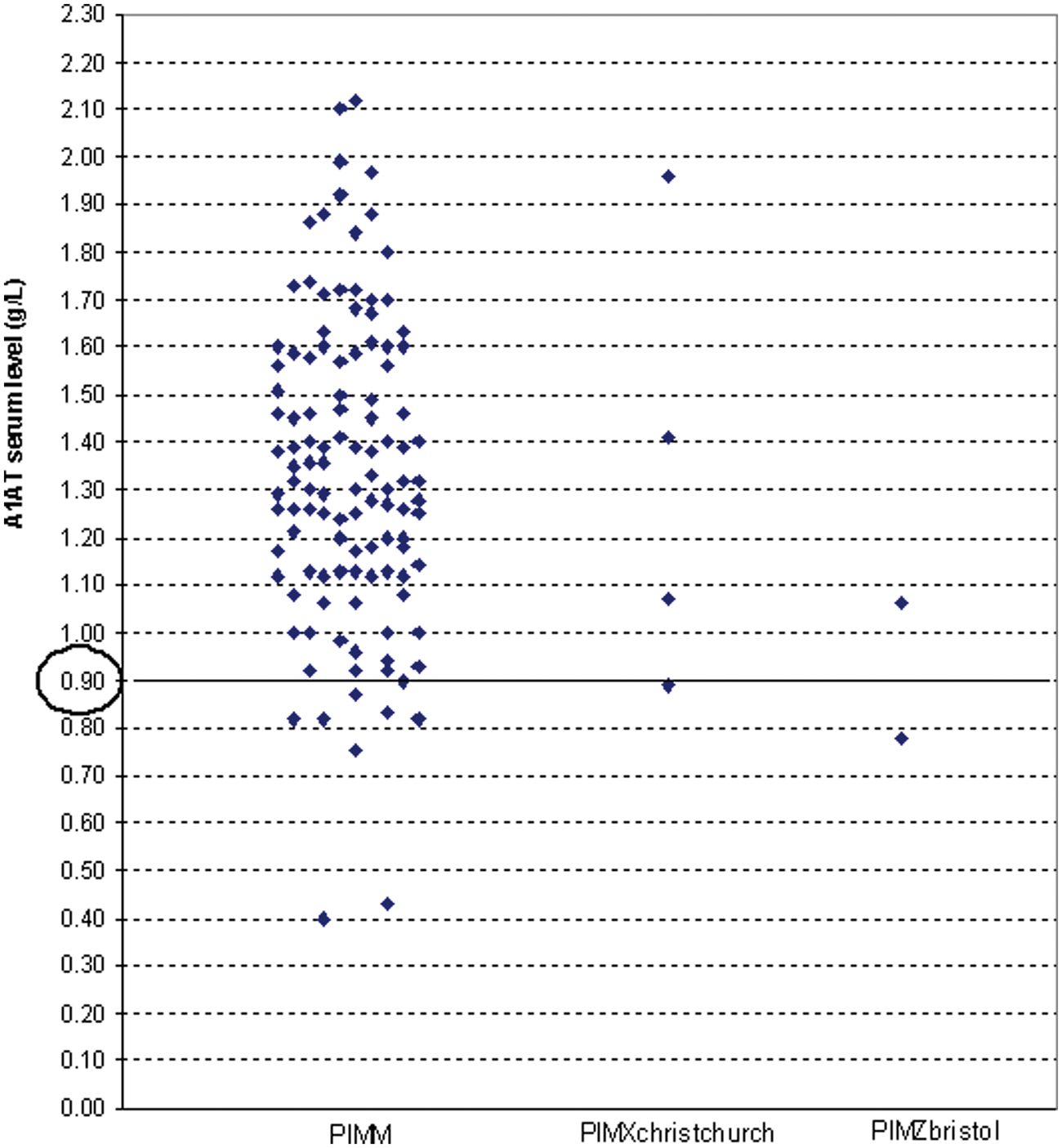
IEF revealed that 124 patients (95.4%) and all of the control subjects (100%) were homozygous for the M allele. Four patients (3.1%) were identified as the heterozygous PI MX phenotype by comparison with control of PI MX phenotype. Two (1.5%) patients had an abnormal pattern with a single intense band that appeared more cathodal than the other five bands of the common M (two intensely stained bands and three minor bands). Comparison with sera control of known phenotype showed that the intense band was cathodal to the Z allele. The phenotypes of these two patients were suggested to be heterozygote PI M? (Figure 2).
The Zbristol variant determined in two children with liver disease. (a) Lane 1: control MZ. Lane 2: heterozygote MZbristol of index 1, A1AT serum level: 0.78 g/L. (b) Lane 1: control MZ. Lane 2: heterozygote MZbristol of index 2, A1AT serum level: 1.06 g/L. Arrow shows the Zbristol band.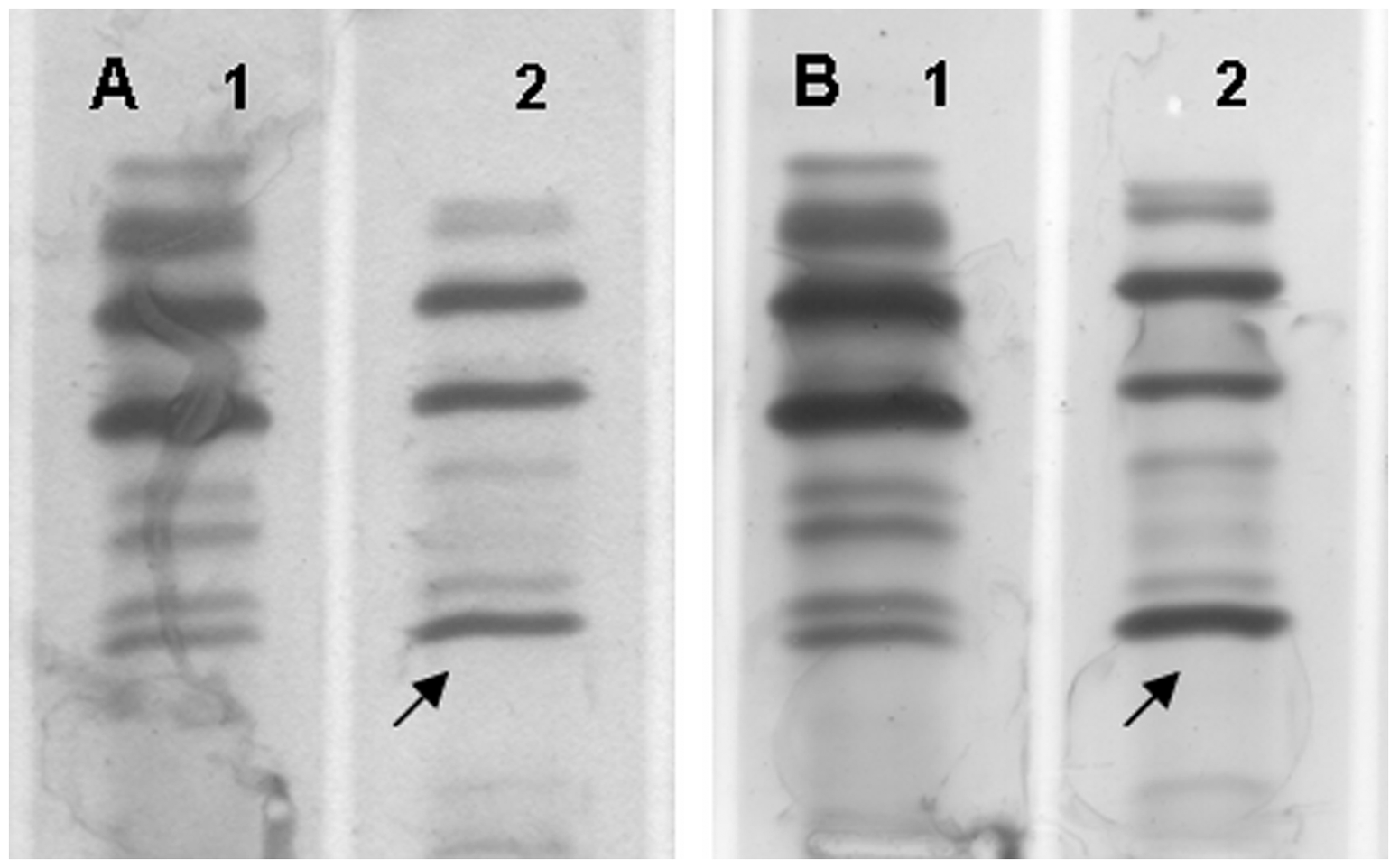
Genotyping was indicated for patients with reduced A1AT serum concentration and/or patients with phenotypes other than PI MM. Patients with normal A1AT serum concentrations and normal phenotypes were randomly selected for genotype to confirm that we had missed no cases of an A1AT deficiency allele. The coding region of SERPINA1 with exons 2 and 5 of the gene was amplified and sequenced. Sequencing results of PCR products from the two patients with the PI M? phenotype revealed a C to T point mutation (g.12781 C>T, SERPINA1; RefSeq NG_008290.1), leading to amino acid change at codon 85 (Thr85Met) of exon 2, which was identified in both patients. Based on the genetic profile, we have identified this as the PI Zbristol mutation
7
(Figure 3). Four other patients that contained a ‘PI MX’ phenotype variant by IEF were identified as PI MXchistchurch genotype. These data were in good agreement with that of sequencing analysis (data not shown).
Exon 2 analysis of index 1 (left) and index 2 (right) shows heterozygote MZbristol genotype. Allele Zbristol differs from the normal PI M allele by the C to T point mutation changing the normal codon for Thr85(A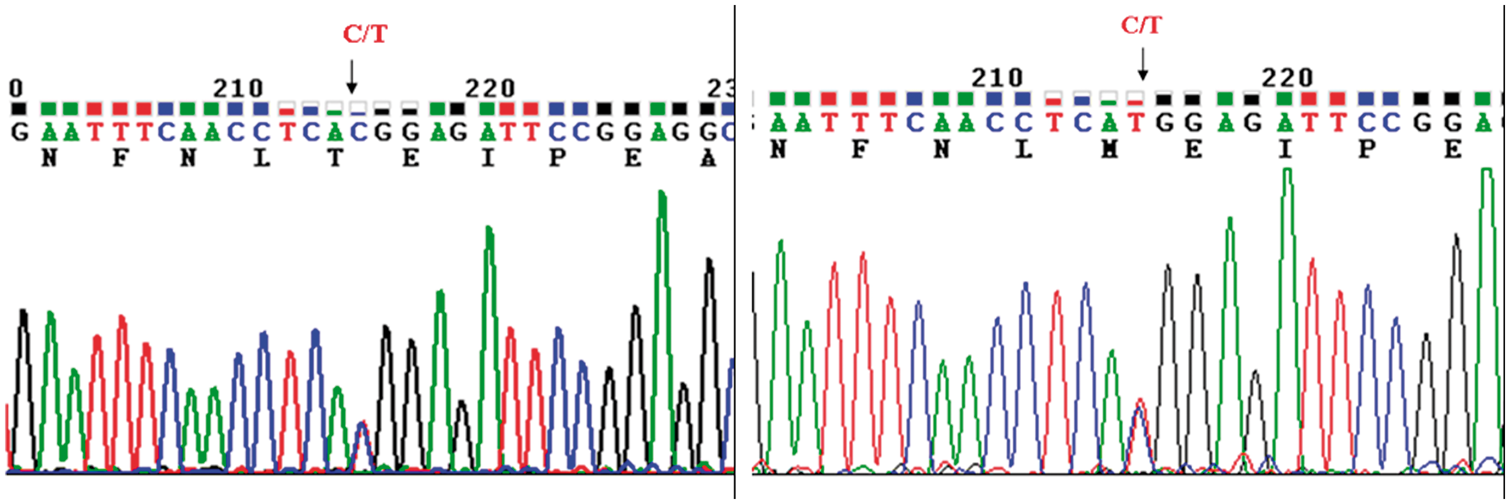
Patient 1 with PI Zbristol was a male infant from Dan Phuong, Ha Noi city, admitted at two months of age and diagnosed as neonatal hepatitis with jaundice revealed since one month of age. Laboratory investigations during hospitalizationrevealed serum A1AT of 0.62 g/L the first time and 0.78 g/L the second time, both lower than normal. The patient had no symptoms of acute inflammation. Moderate hyperbilirubinaemia with 84 µmol/L of conjugated bilirubin and elevated liver enzyme activity were documented with Aspartate aminotransferase – AST (217 IU/L, or 3.6 × upper reference limit for age [URL]), Alanine aminotransferase – ALT (116 IU/L, or 2.3 × URL), Alkaline phosphatase – ALP (911 IU/L, or 2.6 × URL), Gamma-glutamyl transpeptidase – GGT (235 IU/L, or 2 × URL). Prognosis was found to be good and the child was thriving normally. Patient 2 was a 10-week-old male infant from Yen Phong, Bac Ninh province, with a serum A1AT of 1.06 g/L. He was diagnosed with undefined neonatal hepatitis. The jaundice revealed soon after birth. Although he had hepatosplenomegaly, no liver failure was detected. Laboratory investigations revealed persistent cholestasis and hepatocellular injury with liver function tests demonstrating: 159 µmol/L of conjugated bilirubin, 421 IU/L (7 × URL) of AST, 240 IU/L (4.8 × URL) of ALT, 811 IU/L (2.3 × URL) of ALP and 1293 IU/L (10.8 × URL) of GGT. Clinical symptoms and laboratory tests reduced slowly. These are the first two cases of a rare A1AT deficiency allele found in Vietnamese patients.
Discussion
In this study, we investigated A1AT variants in paediatric patients with neonatal cholestasis and other chronic liver diseases. Of the total 130 liver disease infants included in this study, a very rare deficient allele for A1AT, named Zbristol, was identified for the first time in two patients who had A1AT serum level of 0.78 g/L (Index patient 1) and 1.06 g/L (Index patient 2). The Zbristol allele was first described in a woman with an obstetric history of three perinatal deaths from fulminant liver disease and no living offspring. She, her father and her son who died at two days were PI M1Zbristol heterozygotes. The other son who died at five weeks of liver failure was PI M and the sister who died was not tested. The Zbristol protein is active as a proteinase inhibitor but appears to be deficient in the plasma to about the same degree as the S protein in MS heterozygotes. The relationship between Zbristol and fulminant liver disease in the offspring was unclear because this variant existed in only one of the perinatal/neonatal deaths from this case. 7 It is likely that our two liver disease infants with Zbristol supports the relationship of this variant with paediatric liver disease.
Although our results showed that 11 (8.5%) patients had a reduced serum level of A1AT below 0.90 g/L, only one of these patients had a deficient allele which was detected by phenotyping and genotyping analysis. By contrast, the cut-off of 0.90 g/L did not detect index patient two with the Zbristol allele. Therefore, serum A1AT concentration has a sensitivity of 0.5 and specificity of 0.92 using the 0.90 g/L cut-off in this study population. Previous studies have also shown that quantification of A1AT serum concentrations alone is not sufficient to diagnose genetic causes of A1AT deficiency. As A1AT is autosomal co-dominant inherited, a deficient heterozygote individual may have a serum A1AT concentration greater than the recommended cut-off. Mutation in the A1AT gene is not the only cause of A1AT deficiency. Secondary causes of A1AT deficiency include liver damage or conditions such as protein-losing enteropathies that cause a general decrease in serum proteins. 8 This occurred in four of 11 suspected A1AT deficient subjects with low serum levels who had severe liver disease at the time of presentation in our study. Hence, using the recommended cut-off of 0.90 g/L, we consider the quantification of A1AT in serum to be of limited value as a screening test for the investigation of primary causes of A1AT deficiency. Therefore, the differential diagnosis of A1AT deficiency to investigate the aetiology of liver disease in paediatric patients should include phenotype and genotype analysis to investigate the A1AT variants. 6,8
IEF is technically challenging because of the complex micro-heterogeneity of the A1AT protein and the difficulties in interpreting of rare alleles. Analytically, PI Zbristol is very unusual because it focuses at a more cathodal position than the Z protein and shows an abnormal pattern of isoforms. This PI Zbristol variant results from a missense mutation in exon 2 at codon 85 occuring on the common M1(Val 213) genetic background, which disrupts the N-glycosylation site starting at Asn 83 preventing glycosylation at residue 83 in the PI Zbristol protein, which explains the protein IEF and SDS gel electrophoresis results. 7 Phenotyping alone cannot distinguish between an individual who is homozygous for a single allele and an individual who is heterozygous for that allele in trans to a null allele. In addition, severely deficient alleles, null alleles, especially deficient M-like alleles cannot be identified. 6,8 For these reasons, recent studies suggested a workup that includes genotyping by direct sequencing of the coding regions of the A1AT gene as a complementary assay in order to identify deficient alleles in susceptible patients. 6 The SERPINA1 gene is organized into four coding exons and three noncoding exons. Most of the deficient alleles associated with liver disease such as Z, Mmalton, Mduarte, Siiyama, I alleles are located in coding exons 2 and 5. 1 Therefore, in our study, only exon 2 and exon 5 were sequenced, based on the A1AT concentrations and phenotyping test results. This is potentially one limitation of our current study as ideally the entire gene may be sequenced to confirm there are no cases of null alleles, which may be the cause of low A1AT concentrations in some patients.
A1AT deficiency (first documented in 1963 by Laurell and Eriksson) is an autosomal co-dominant condition common among Caucasians. In contrast to the high prevalence of A1AT deficiency in the Caucasian population, this disorder is rare among Orientals. 9,10 The most common deficiency allele is PI Z (Glu342Lys), and 95% of individuals with A1AT deficiency are of PI ZZ type. Other mutant alleles, including null and dysfunctional alleles, are relatively rare and occur in approximately 5% of individuals with A1AT deficiency. 6 Previous studies have shown a low frequency of the Z allele in Asian countries. 11,12 Instead of the Z allele, other deficiency alleles were reported. QOhongkong (a null variant) and PI Pisttburgh (Met358Arg) (a dysfunction allele) were reported among Chinese. Siiyama (Ser53Phe) (a deficiency allele), in conjunction with Mnichinan, Mmalton and QOclayton was reported among Japanese. 13 –15 In our study, the first two cases of A1AT Zbristol deficiency were noted in Vietnamese paediatric patients.
Conclusion
In summary, A1AT deficiency is widespread throughout the world, with a high prevalence in Western countries. It also is clear that A1AT deficiency is not just a disease of Caucasians, but is prevalent in many different races throughout the world, which now includes Viet Nam. We recommend that the laboratory diagnosis of A1AT deficiency including A1AT concentration determination, phenotype and genotype testing should form part of the routine differential diagnosis of paediatric liver disease of indeterminate aetiology in Vietnamese patients.
Footnotes
Acknowledgements
We would like to express our gratitude to Dr Ann Read, Dr James Doery and Mr George Streitberg for helping us set up the IEF technique and providing us control samples. We also thank Dr Ronda Greaves (School of Medical Sciences RMIT University) for carefully reviewing this manuscript.
Declaration of conflicting interests
None of the authors have any competing interests.
Funding
The study was supported by a grant from the Service of Science and Technology of Ha Noi (Code 01 C-08) for phenotyping and determination of A1AT levels.
Ethical approval
The study was approved by committees of the Service of Health of Ha Noi and the Service of Science and Technology of Ha Noi, Ref 01 C-08/07-2009-2.
Guarantor
HTH.
Contributorship
HTH: Conceived, designed and performed the experiments. PTN, NGK and LQH: Instructed and supervised. LQH: Contributed reagents/materials/analysis tools. HTH: Wrote the paper. All authors read and approved the final version.