
Abstract
Substantial advances occurred in phlebological practice in the last two decades. With the use of modern diagnostic equipment, the patients’ venous hemodynamics can be examined in detail in everyday practice. Application of venous segments for arterial bypasses motivated studies on the effect of hemodynamic load on the venous wall. New animal models have been developed to study hemodynamic effects on the venous system. In vivo and in vitro studies revealed cellular phase transitions of venous endothelial, smooth muscle, and fibroblastic cells and changes in connective tissue composition, under hemodynamic load and at different locations of the chronically diseased venous system. This review is an attempt to integrate our knowledge from epidemiology, paleoanthropology and anthropology, clinical and experimental hemodynamic studies, histology, cell physiology, cell pathology, and molecular biology on the complex pathomechanism of this frequent disease. Our conclusion is that the disease is initiated by limited genetic adaptation of mankind not to bipedalism but to bipedalism in the unmoving standing or sitting position. In the course of the disease several pathologic vicious circles emerge, sustained venous hypertension inducing cellular phase transitions, chronic wall inflammation, apoptosis of cells, pathologic dilation, and valvular damage which, in turn, further aggravate the venous hypertension.
Introduction
Lower extremity chronic venous disease is a pathologic condition characterized by venous varicosity and with venous insufficiency in its more advanced forms. It is a typical civilization disease: low prevalences of preindustrial societies massively increase with their Westernization.1–3 It affects around a quarter of the population of the developed world during his/her lifespan at the level causing clinical problems. Causes loss of workdays, limited work activity and effectivity, health care costs amounting to billions of dollars yearly, much human suffering, even death, substantial aesthetic hindrance, inconvenience, and disability in hundreds of millions human subjects. Cosmetic problems may be encountered in up to three quarters of the general population. Several national statistics prove the sensitivity of all mankind to this disease with moderate geographical and racial differences.4–17
Classical view attributes the frequent lower body venous problems in humans to late development of bipedalism and erect body position. However, powerful adaptive processes have been developed to encounter the increased pressure of the venous blood column induced by gravitation. 18 Historical time for a successful genetic adaptation seems to be more than earlier expected. Zoological observations prove that primates, especially apes spend a substantial time in a somewhat erect body position. Chimpanzees and gorillas frequently move around with an erect upper body, walking on two legs, occasionally assisting their balance with the knuckles of their hands.19,20 New anthropological findings, with proven isotopic dating and biomechanical analysis of footprints date back the appearance of erect walking in early anthropoids to very early times (>3.5 million years ago).21–23 Scattered publications on people living under natural conditions with much time spent with walking and running show that they are surprisingly devoid of leg varicosity problems.24–26
The past few years brought substantial advancement both in the clinical-pathological and research areas. High resolution and Doppler ultrasonographies are routinely used,27,28 laser illumination of superficial veins is widely available, 29 and in some institutions even near infrared imagination can now be applied to investigate the pathological venous network.30–32 A substantial amount of data has accumulated regarding the network and segmental deformations, and altered flow patterns in varicose legs. Broad use of intravenous ablation, minimally invasive surgery, and careful documentation of the outcomes accumulated substantial practical knowledge with much scientific value.33,34 A majority of clinicians are now convinced that incompetent perforator veins should be in the background of practically all varicose deformations.27,35 That flow is an important component in the development of varicose deformations has been confirmed in a series of animal experiments in our laboratory.36–38
Pathological and experimental studies on human and animal tissues and cells revealed new physiological, cellular, biochemical, and molecular biological factors with essential roles in the pathomechanism of this disease.39–41 Some emerging contradictions in the literature regarding cellular pathology processes, we are convinced, can be explained by the typical heterogeneity of the lesions.39,42 What is more, the sequence of events during propagation of the disease can be better understood if we are aware that these heterogeneities many cases reflect the different phases of the disease at different locations of the venous network. 43 Detailed description of hemodynamic and cellular pathology events at different sites of the diseased network, while being in different phases of disease development, will provide more guidance for prevention and prognosis, for indication, selection, and execution of surgical and intravascular interventions and for postoperative treatments. Better knowledge of cell pathology processes perspectivically opens new possibilities for more effective pharmacological interventions. Further, several similarities between the general aging process and local varicose venous wall remodeling have been demonstrated.44–47
Our aim was to overview the pathomechanism of this disease integrating both classical and emerging new knowledge in the clinical, pathological, and scientific fields. We included published material describing important steps and details of the complicated pathomechanism. If some contradiction does exist (as in the case of apoptotic processes), we attempted to provide a full picture. Also, cellular physiological details have been described, the full significance of which in the pathomechanism of the chronic venous disease is still not clear, but it can be expected based on analogies in other areas of medical sciences (e.g., epigenetic changes).
Epidemiology
Varicosity disease is very common in modern society. In the Edinburgh general survey in 80% of males and in 85% of females telangiectasias and reticular veins could be found in the 18–64°years old population, while explicit varicosities were observed in 40% and 16%, respectively.5,14,15,46 Similar observations were made elsewhere.4,6–10,12,16,48–50
Sex and Lifestyle
Clinical symptoms and complaints have been found to be more frequent in females in several studies 5 but in age matched samples males produce more serious symptoms. 46 The Framingham study provided statistical proof that persons, both males and females, spending more than 8 hours daily with sedentary activities (standing or sitting) had significantly higher incidence of varicose veins than subjects with less than 4 hours. 44 Work activity in the standing and even in the continuously seated body position increases the frequency of lower extremity varicosities.6,9,50–62 Increased body weight63,64 and wearing high-heeled shoes 65 also form increased risks. With accelerated urbanization of sub-Saharan Africa, varicosity appeared in high numbers in an earlier unaffected population. 66
Genetics, Changes in DNA, and Epigenetics
Family occurrence is characteristic for this disease.67,68 Swedish hospital statistics have shown a substantial familial occurrence, the involvement of spouses, however, proves an additional marked life-style component. 69 Comparison of the inter-twin correlation of the in vivo mechanical properties of the femoral vein of monozygotic and dizygotic twins demonstrated a substantial (40–60%) genetic inheritance. 70 Proneness for the disease has a substantial genetic, inherited component.71–77 Identification of those minor genetic variabilities that might determine the vulnerability to venous disease are just now gaining momentum.67,74,78–87
In 2012, Anwar et al. 78 identified nine mutations associated with varicose vein development and six further genetic variabilities which tend to promote venous ulcer progression. They include such diverse traits as Ehlers-Danlos syndrome (affecting the COL3A1 gene causing connective tissue weakness), defects of the von Hippel-Lindau gene (HIF1α regulator, angiomas), the FOX2 gene (gene of the Forkhead box P2 transcription factor protein, responsible for lymphatic endothelium and venous valve development; see the sections below), and genes of thrombomodulin (endothelial antithrombin effects), desmuslin (intracellular intermediate filament protein, smooth muscle cytoskeleton scaffold), and the MTFR (methylene tetrahydrofolate reductase, hyperhomocysteinemia) gene. FOXC2 and GJC2 (connexin47) mutations have been identified as inducing vulnerability to varicosity. 88 An autosomal dominant variation of the FOXC2 gene has been found to be connected to a familial form of varicosity. 89 This may be explained by the role these genes play in the control of venous valve development (See Chapter above “Genetics, Changes in DNA, and Epigenetics”). In a recent work by Fukaya et al., 75 genetic constitution of half a million people was tested with the GWAS (genome wide association) technique, and 855 single nucleotide polymorphisms were identified as statistically connected to lower extremity varicosity. Thirty genetic loci can be considered robustly associated with varicosity. Three of them are: the CASZ1 gene (Castor Zinc Finger 1, located in a known blood pressure determining locus), a variant located in the neighborhood of the vascular mechanosensitive PIEZO1 locus, and another in the neighborhood of the GALNS (galactosylamine sulfatase, polysaccharide chain synthesis) structure gene. Dysregulated expression of the CASZ1 gene in varicose specimens has also been confirmed in a recent work, 90 together with WDR92 (caspase3, an apoptosis regulator) and RSPO3 (planar cell polarity). Decreased expressions were confirmed for CASZ1 and WDR92 and also for such diverse genes as LIMA (actin filament crosslinking), ABCB10 (mitochondrial ABC transporter), DNAJC7, the gene of HSP40, a heat shock protein (co-chaperone function, correction of protein steric structure), C1S (major constituent of complement C1), and CXCL1 (chemokine, neutrophile chemoattractant). Enhanced expressions for PHLDA1 (apoptotic regulator) and SERPINE1 (fibrinolysis inhibitor) have also been confirmed. On the other hand, single nucleotide polymorphisms in the genes of 13 inflammatory factors did not show association with varicosity in a Russian population 91 while an association with the MHC class III genes, genes and areas controlling vascular development and remodeling could be established.92,93
In a Chinese population, the −1562C allele variation in the MMP-9 (matrix metalloproteinase type 9) gene was identified frequently in varicosity patients, and the involvement of the promoter region of the TIMP-2 (tissue inhibitor of metalloproteinase) gene was also suspected. 87 In a European population, in addition to variations in TIMP-2, variations of MMP-2, MMP-9, and MMP-12 were found to be associated with higher risks. 94 The involvement of the variability of the DYSPL2 gene (coated vesicle formation) may be surprising, but more understandable is the role of the VSTM2L (cell–cell adhesion) in a Chinese population 84 ; and even more explainable are the involvements found for EFEMP1 (fibulin like extracellular glycoprotein) and KCNH8 (pore forming subunit of a potassium channel) in a German population. 80 Analysis of data provided by close to 100,000 23andMe customers (commercial genetic heritage tests) revealed association of self-declared varicosity with the ABO blood group, and among others also PIEZO1, VEGFA, and CASZ1, partially confirming the previous observations. 79 Summarizing the literature up to 2014, Bharath 95 lists several further genes variance of which appears to be connected with varicosity: COL1A2 (part of collagen type 1 alpha molecule), FOXC2 (forkhead transcription factor C2; see also the Valvular Damage as well as the Smooth Muscle and Contractility chapters), HSP90 (heat shock protein 90), ILK (integrin linked kinase, signalization through cell surface contact molecules), MGP (matrix Gla protein calcification?), Oct-1 (octamer binding transcription factor 1), TGF-β1 (transforming growth factor type 1 beta), collagen types I and III, and VEGF-A and VEGF-R (vascular endothelial growth factors types A and R). In an Italian set of studies on venous ulcers (more serious form of the chronic venous disease), the involvement of the HFE gene (hemochromatosis gene, iron uptake by cells), FPN1 (ferroportin, nonheme iron removal from cells), and of the more plausible MMP-12 (matrix metalloproteinase type 12) could be identified. 81 Single nucleotide polymorphisms of the FGFR2 (fibroblast growth factor receptor type 2) may also be associated with these more serious forms of the disease. 86
We can conclude that the complex and sensitive machinery of building and maintenance of leg venous networks including the ability to fulfill its function under markedly variable hemodynamic conditions (flow and pressure) can be subjected to genetic damage at very diverse points. Genetic tests (DNA sequence analyses) do not form a routine examination step of chronic venous disease patients these days. However, in some serious familial forms of the disease, such tests may yield therapeutically useful information.
An important question is whether local DNA damage can contribute to the pathomechanism of pathological wall remodeling observed in chronic venous disease? It seems that in addition to age-induced DNA mosaicism and DNA breaks in varicose tissue the level of DNA damage is enhanced. This is demonstrated by the elevated expression of the single-strand DNA break detecting protein PARP. 96 Damage to the mitochondrial DNA could be directly proven. 97
According to modern observations, non-coding RNA fragments can interact with nucleic acids and influence replication, transcription, and translation processes.98,99 The expression pattern of circRNAs of varicose saphenous veins were significantly altered, with 232 different circRNAs aberrantly expressed. These affected the functions of microRNAs regulating among others catabolic processes, cytoplasm and ATP binding as well as components of the NF-κB (nuclear factor κB, chronic inflammation) signalization pathway. 100
Complexity of the genetics of the disease was well demonstrated by the strong statistical correlation in a Chinese population between venous insufficiency and a seven nucleic acid base pair insertion/deletion polymorphism in the 3′ untranslated region of the COL1A2 (α2-type I collagen gene), supposedly affecting the association and control of the synthesized mRNA with a specific microRNA. 83 A third group of non-coding RNAs also seems to have pathological significance: the expression of lncRNA-GAS5 (a long noncoding RNA, inhibitor of tumor cell apoptosis with mTOR-mediated proliferation effects) is decreased in great saphenous veins affected by varicosity. 99 Further studies are needed to reveal whether this is an essential step in the pathomechanism or just a side-effect of other pathologically more important processes.
Regarding the future, it is promising that inherited genetic programs defining a venous type of endothelial cell differentiation (in contrast to arterial) have been successfully identified: in venous endothelial cells the ephrin receptors EphB3 and EphB4 are expressed. 101 Also, genes regulating venous valve development have been spotted, might be surprising that they show a certain overlap with genes involved in lymphatic vessel differentiation. These genes are the ephrin-B2, Prox-1, Vgfr3 (fibroblast growth factor receptor 3), integrin-α, 102 and FOXC2. 85 No surprise that we can find them among genes whose minor genetic variations can be connected to varicosity (see above).
Pathological Hemodynamics of the Leg
Hemodynamic factors initiate, maintain, and aggravate the chronic venous disease.
Basic hemodynamics of the lower extremity veins with deep and superficial and perforating veins and normal direction of flows has correctly been described in 1929 by McPheeters. 103 Even ligation of perforators was suggested as early as in 1938 by Linton. 104 Advanced instrumentation now allows us to observe directly that blood flow in healthy perforators is very limited at rest and during isometric skeletal muscle contraction, but there is strictly unidirectional flow (from superficial to deep veins) during intermittent calf muscle contraction. 105 Identification of reverse flow in incompetent perforator veins is now part of the everyday clinical practice.28,106–118
Some earlier theories suspecting primary role of pathologic alterations in the venous wall itself have now few followers,119,120 these do not command wide support of specialists with large experience in patient care. Nevertheless, we have to admit that the aging processes of the wall are in many respects similar to those of varicosity, and they are accelerating the pathologic developments there.47,121 Further, genetic constitution of the tissues forming the venous wall is also an important pathogenic component (see above chapter “Genetics, Changes in DNA, and Epigenetics”). During the whole course of the disease, pathologic hemodynamics as well as pathologic wall/valve remodeling progress hand-in-hand. 35 They amplify each other mutually: irreversible positive feedback control circuits emerge. 122 No surprise, causal connections will be found in both directions. Fortunately, at the same time, this also gives possibilities for successful therapeutic intervention to disrupt these vicious circles along both lines.
Classically, sustained elevated venous pressure had been thought to induce and maintain varicose deformations, with valve incompetence in the main branch of the saphenous vein, close to its confluence with the deep femoral vein emerging in advanced lesions only. Incompetence of the perforating branches as a significant initiating factor in the varicosity was suspected first by Moore in 1951 123 and in 1953 by Cockett, 124 later proven ultrasonographically by Lawrence in 1977. 116 With the availability of improved ultrasonographic and physical examination techniques in more and more varicosity cases, the presence of incompetent perforant veins could be identified. These feed the malformations of the superficial venous system leading to volume overburden, elevated venous pressure, and irregular and turbulent flow.27,28,35,106,107,110–115,125–127 Extreme views, that no varicosity process is without incompetent perforators have been published, but most specialists now hold the somewhat more conservative opinion that reverse perforator flow can be spotted in majority of varicosity cases.107,108,128 Few deny the significance of incompetent perforators. 109 Valvular incompetency appears in several superficial, deep, and perforating veins,28,106 their ultrasonographically observable prevalence increases with the increasing severity (higher grade of C during CEAP classification) of the disease.107,111,117,128
Regarding the two basic hemodynamic factors, transmural pressure and luminal flow, usually the role of pressure is emphasized. Elevated leg venous pressure is the mainstay of the disease. Morphological dilations are expected to emerge as the result of sustained, pathologically high venous pressure maintained by gravitation, 129 valve incompetence in the affected vein, 130 as well as volume load through incompetent valves elsewhere. 131 High venous blood pressure induces elevated circumferential wall stress, 132 damages the endothelium, activates inflammatory cascades in the wall, initiates expression changes and migratory transformation of smooth muscle cells in the media and finally their apoptosis, and leads to serious fibrosis of the wall and deformation of the lumen. 131 Expression of several proteins will be enhanced, such as vascular endothelial growth factor, endothelin-1, interleukin-6, vascular cell adhesion molecule, intercellular adhesion molecule, matrix metalloproteinase-2 and -9, and plasminogen activator inhibitor-1, orchestrating the pathological remodeling of the venous wall 133 (see “Intima and Endothelium,” “Smooth Muscle and Contractility,” “Extracellular Matrix” as well as “White Blood Cells, Inflammation, and Metabolism” chapters).
The other basic hemodynamic factor, venous flow is also an important component of the pathogenesis. Its role is not restricted to the induction of volume overburden and thus elevated pressure. Polysaccharide chains at the luminal surface of (intact) endothelial cells are able to sense endothelial shear and adjust lumen diameter acutely by initiating cellular signal pathways to release the vasodilator agent NO and relaxing smooth muscle. Such mechanisms are present also at the venous side of the circulation. 134 In addition, general angiological evidence proves that morphological lumen diameter of a vessel on the long run is controlled by the flow inside its lumen: higher flow will result increased morphological lumen. Sustained flow changes initiate morphological remodeling of the wall. This flow-induced morphological remodeling of lumen diameter is present in veins.135,136 In analogy to arteries, in the case of veins, we can also expect continuously, slowly working physiological wall remodeling processes that adjust the morphological lumen to luminal flow and adjust the thickness, collagen, elastin, and contractile protein content to transmural pressure load.137–139 Animal experiments performed in our laboratory demonstrated that during the flow-induced remodeling process, monocyte infiltration helps remove the old structures, and accelerated cell division produces cells whose contractile protein content and elastin expression is transiently insufficient: the mechanical strength of the wall is weakened. While stable, healthy veins are surprisingly resistant to higher pressures,136,140 elevated gravitational load can induce varicose changes in the venous system of the rat being in the process of flow remodeling. 37 Surprisingly high blood flow values could be measured in human skin telangiectasias,141,142 suggesting the presence of flow-induced remodeling processes in these cases. Because of the disturbed morphology of the network, at other places stasis can be present, 142 inducing alternative wall remodeling processes. A theoretical work even suggests that some flow obliteration and redirection toward less adapted venous segments should be in the background of all leg venous pathologies. 143 Flow overload of remaining perforators can explain some of the recurrences after ablation. 130 The significant role of turbulent flow in formation of the morphological deformities of the superficial venous system has been recognized by Fegan. 125
In addition to pathologic feedbacks between hemodynamics and wall damage, there is positive feedback between the pathological hemodynamic elements themselves: incompetency of superficial veins frequently associates with incompetency of perforators and vice versa. 112
Furthermore, disturbed hemodynamics are responsible for the most serious consequences of the disease. If elevated venous pressures are retrogradely transferred to the microcirculation, edema, pain, reduced tissue blood flow, and inadequate tissue oxygenation will be the result with symptoms of skin atrophy and even necrosis: treatment resistant ulcers can emerge. 144 To stress the clinical significance of such lesions, many phlebologists prefer the “lower extremity chronic venous disease” expression for the disease complex.
The International Union of Phlebology consensus statement of year 2016 contains a detailed description of the hemodynamic changes found in lower limb chronic venous disease. 33
Gravitational Effects
Gravitational effect is an essential component of initiation, maintenance, and progression of chronic venous disease of the lower extremity. The up to 80–100 mmHg hydrostatic venous pressure at the level of the dorsal pedis vein in the standing body position is easily eliminated by rhythmic movement of the calf muscles (muscle pump) if the valves are intact. Continuous isometric skeletal muscle contraction or damaged valves will result in lasting elevated venous pressures.129,140,145–150
The healthy venous wall is surprisingly resistant to pressure elevations. We kept rats in tube cages tilted by 45° which elevated saphenous vein pressure from about 5 to 10 mmHg in a chronic manner. A slight morphological dilation of the main branch and its tributaries,18,135 hyperpolarization of vascular smooth muscle cells, 151 increased sympathetic innervation of the wall, 152 increased myogenic tone, 153 and degranulation of endothelial cells 154 were observed. Explicit varicosities or telangiectasias did not develop. Comparing human small veins of similar diameters from the neck area and from the leg, leg veins had significantly thicker walls, more elastin, and more contractile protein expressed in their walls. 155 Secretory granules in the endothelial cells resembled more the elongated structure of the Weibel–Palade bodies. 156 This proves that physiological remodeling processes are present in the venous wall to control wall thickness and histological composition by distending forces on the wall. These aim to stabilize wall stress and stress on circumferential force-bearing histological components, similarly, as described by Folkow and Rodbard for arteries.137–139 In veins being in the process of flow-induced morphological remodeling, which is accompanied by degradation of the old force-bearing elements, increased rate of cell division, slow accumulation of newly expressed contractile and connective tissue proteins, the sensitivity to gravitational load increase. Real varicose deformations could be produced in lower limbs of rats, when development of bypassing collateral vessels was initiated by narrowing the main branch of the rat saphenous vein and then exposing these vulnerable new veins to gravitational load by keeping the animals in tilted cages. 37
Muscle Pump
In the standing body position, if the venous valves are intact, rhythmic activity of the leg muscle can induce a 50–70 mmHg drop in the supramalleolar venous pressure.40,145–150 During walking and running, surprisingly high pressures (180–270 mmHg, with substantial scatter) were measured in human volunteers in the soleus muscle, compressing the deep veins. 157 Modern ultrasonography makes it possible to identify a high number of perforators in the leg venous system. Doppler observations demonstrate that in most normal subjects, there is no flow at all at rest in the perforators, while unidirectional flow develops during exercise. Perforator flows reach 80 mL/min per leg. 105 A successive activation of leg muscles in proper order during walking is needed for the effective work of the muscle pump.158–161 Muscles that support the standing body position and those ensuring intermittent muscle contraction for walking may be partly different, this appears to present problems for the leg muscle pump. 162 Effectivity of the calf muscle pump is dependent on the type of shoe worn by the patient, high-heeled shoes have been described to be disadvantageous in this regard.65,163 With electrical stimulation of the peroneal nerve, 17–36% elevation of the volume flow could be achieved in the main deep veins of the calf. 164
Valvular Damage
Healthy venous valves are delicate structures, formed by a thin connective tissue layer between two endothelial sheaths. 118 Upon proximal pressure elevation, decreasing flow velocity and vortex flow into the sinuses induce closure without any reverse flow.165,166 In many cases, venous valves are located in side branches just prior to their confluence with a larger branch.167–169 Valves in smaller veins are distributed in a more random manner. 170 According to our observation, this will induce a tortuous route in developing from small veins collateral networks.37,136 At the site of venous valves, we can usually observe a moderate physiological dilation of the lumen. More significant dilations promote valvular incompetence.35,171 Perforator veins with diameters over 3.9 mm 112 or 3.5 mm 171 are mostly incompetent. Venous valves are surprisingly resistant against acute higher pressures, but chronically elevated pressures induce local dilation and valvular damage with incompetence. 169
Valvular incompetence of the superficial, perforator, and deep vein valves can be observed in the chronic venous disease accompanying superficial varicose lesions.107,109–116,118,123,166,172 Incompetent valves can be recognized by direct static ultrasonographic investigation based on the incorrect position of the valves or by Doppler investigation by the appearance of reverse flow upon proximal pressure elevation.27,110,173,174
Both pathological and clinical observation of incompetent valves reveal the local morphological dilation of the encasing venous section, the reduced lengths of the cusps, damaged endothelial cover and thickening, fibrosis, and corrugated, uneven valvular leaflet edges (interfering with mobility and exact alignment of the cusps). Thrombocyte aggregation is often present in the valvular sinuses.146,175,176 There is a characteristic inflammatory component in valvular damage. 177 Thickening of the cusps is inversely connected to their proper function. There is doubling of cusp thickness with age, 178 while according to Mouton cusp, thickness depends mostly on pathology in phase C2 clinical class according to CEAP classification. 179
Histological examination of incompetent valves reveals monocytic infiltration. 177 Thickening of the cusps is formed by smooth muscle cells at the base and by collagen at the edge of the cusps. 172 The expression of inflammatory markers IL-6, IL-8 (interleukin type 6 and 8), TGFbeta (transforming growth factor beta), and VCAM-1 (vascular cell adhesion molecule type 1) are elevated in the cusps of incompetent valves. 180 In structures adjacent to incompetent (experimental) valves, T cell, granulocyte, and macrophage infiltration, elevated expression of the chronic intracellular inflammatory signal protein NFκB (nuclear factor kappaB) demonstrated the presence of inflammatory processes in the cusps’ encasement wall. Elevated endothelial adhesion molecules, P-selectin, and ICAM-1 (intercellular adhesion molecule type 1) paved the way for white cell migration. Matrix metalloproteinase types 2 and 9 (MMP2 and MMP9) were elevated showing accelerated degradation of the existing connective tissue structure.181–184 Disruption of the smooth muscle bundles by connective tissue, fragmentation of elastica, and intimal thickening were typical further alterations. 185
There is a positive feedback connection between valvular damage and varicose dilation. 186 Elevated venous pressure damages both the annulus and the cusps. 172 The valves are strong: in animal experiments, surprisingly high venous pressure levels are needed to achieve valvular damage. 187 The number of apoptotic cells is increased in the wall of veins subjected to chronically elevated pressures.183,184
Baron 71 suspected inherited weakness of the valves as a main factor in the pathomechanism of varicosity. A promising perspective is that genes activated during valvular genesis have been lately identified. The expression of integrin-alfa9 and Ephrin-B2 at the site of valvulogenesis is essential for venous valve development and maintenance. Integrin-alfa9 and its ligand Fn-EIIIA are thought to organize the assembly of fibronectin and of the extracellular matrix core. Expression of ephrin-B2 by venous valvular endothelial cells may be surprising, as this protein is known to regulate arterial endothelial identity. Other proteins expressed are prospero-related homeobox1 (Prox1) and Vgfr3 (gene of vascular growth factor receptor type 3), known regulators of lymphangiogenesis. 102 Many patients with mutations in the forkhead box protein C2 (FOXC2) gene have damaged lymphatic vessels, but even those without clinical lymphatic disease have long saphenous vein reflux which supports a role of this gene in the genesis of venous valves. 85 This latter protein cooperates with Vgfr3 (gene of vascular growth factor receptor type 3) and governs epithelial/mesenchymal transitions induced by TGFbeta (transforming growth factor beta). Expression of the PDGF (platelet-derived growth factor) receptor beta is an important step in this transition. 188 Venous valve formation in mice is organized by a ring of specialized endothelial cells, expressing the transcription factor PROX1 and the connexin CX47 (gap junction protein), 88 connexin CX37, 189 and several other markers in a sterically regular manner. Another angiogenetic receptor system also appears to be involved in valvular damage: the expression of the angiopoietin receptor Tie1 (member of the tyrosine-kinase receptor family) diminishes. 190
Chronic venous disease is more frequent in non-thrombotic thrombophilic patients, 191 which demonstrates the significance of thrombotic processes in valvular damage. As an additional connection, the increased in vivo rigidity of the common femoral vein has been demonstrated in such subjects. 192
Valvular Incompetence and Reverse Flow
Moore 123 and Cockett 124 recognized first that valvular defects in non-superficial veins form a decisive component in the pathomechanism of superficial vein varicosity. As we have mentioned, according to modern view, in the majority of varicosity cases incompetent perforator(s) feeding the deformed superficial network can be identified. Reverse flow can be present in the perforators, in the superficial and in the deep veins. Reverse flow can be easily identified with color Doppler when forward and reverse flows are marked with alternative colors.27,28,107,112–117,186,193–195 As the disease progresses, several recirculation circles may be present concurrently. 196 Modern treatment of extensive varicosities includes obliteration of incompetent perforators.130,194,195,197–199
Venous Insufficiency
The above-described processes can develop into venous insufficiency. In cases of venous insufficiency, substantial persistent ambulatory venous hypertension, compromised microcirculation, and risk of ulcer development occur.108,147 Mechanism of the high-pressure valvular damage has been reviewed by Bergan. 131 As an animal model of venous insufficiency, very high, 94 ± 9 mmHg (from 11 ± 2 mmHg) leg venous pressures were established in rats by creating an arteriovenous fistula. Such high pressures increased lumen diameter at the site of the valves while it decreased the length of the cusps and a substantial reflux developed progressively increasing up to 42 days forming a vicious circle. 200 Number of granulocytes, macrophages, T lymphocytes, apoptotic Tunel positive cell,s and the expression of P-selectin as well as of ICAM-1 increased in the affected cusps’ wall.182,183
The hemodynamic interactions during the progression of lower extremity chronic venous disease are shown in Figure 1. We can see that such interactions can form numerous (several hundreds) pathologic positive-feedback (“vicious”) circles. Pathomechanism of lower extremity chronic venous disease. Hemodynamics. Anatomical sites are organized into vertical columns and pathological alterations into horizontal rows. Red arrows, positive effects. Note numerous potential positive feed-back circuits. Blue arrows, negative feed-backs. G, targets of known genetic effects. Clinically observable symptoms are color-coded and named in the bottom row.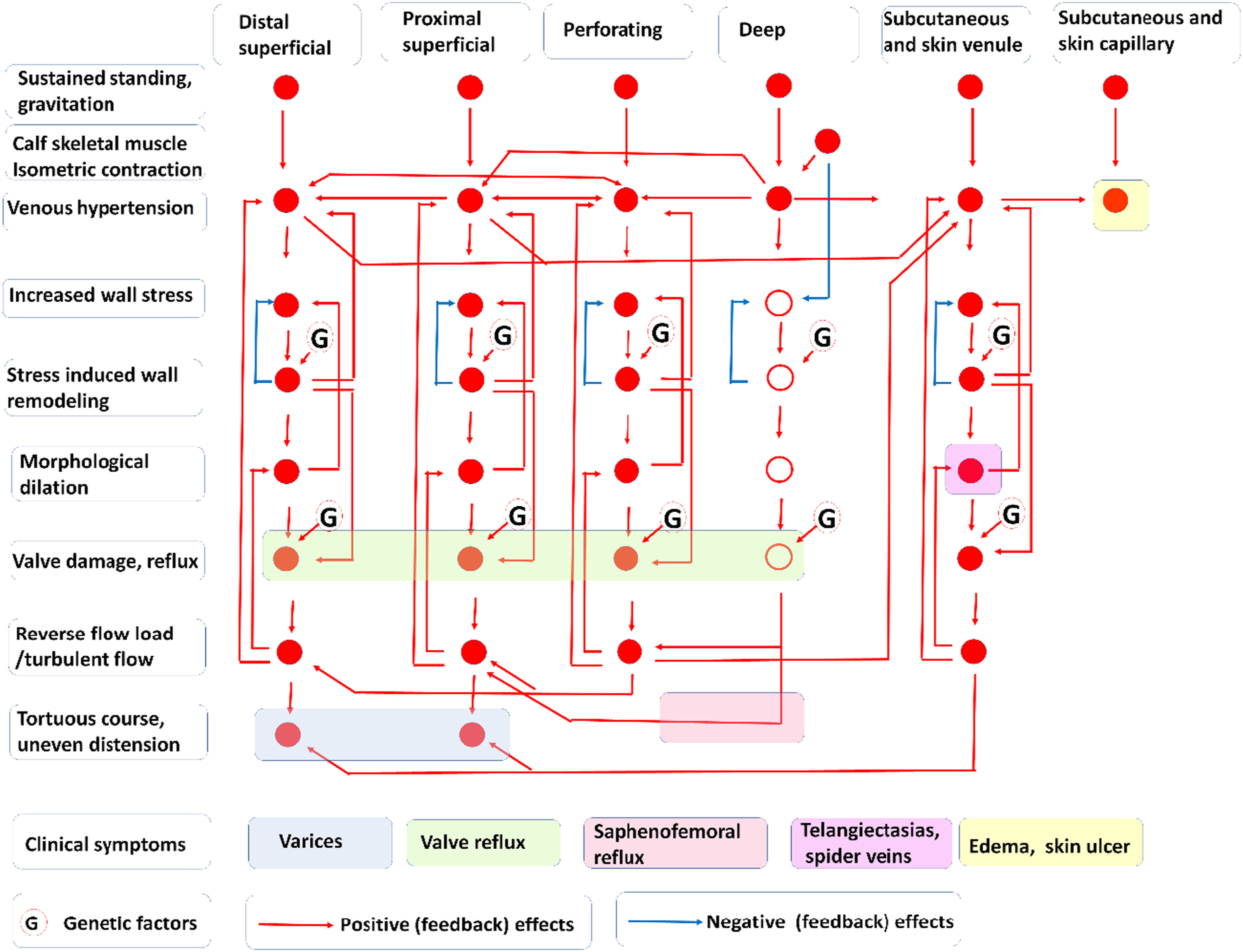
Intima and Endothelium
The intima is a significant element in all chronic vascular diseases.201,202 In its intact form, it prevents large molecular mass components, such as fibrinogen, antibodies, plasma proteins, and lipoproteins to pass from the blood into the vascular wall.203,204 Proper molecular composition of intraluminal surface proteins and polysaccharides prevents platelet and white cell adhesion, and their migration into the wall. All these effects are present in the venous wall.205,206 Intact venous endothelium also produces NO134,207 and prostacyclin 208 with substantial platelet anti-aggregation and vasodilatory effects. Endothelial shear is sensed by polysaccharide molecular chains located at the luminal surface; this induces acute vasodilation through activation of NO-synthase. 209 In case of a chronic effect, the endothelium adjusts the morphological lumen to flow by mechanisms not yet fully understood.210–212 One component of the shear force sensing pathway is Piezo1, a mechanosensitive plasma membrane Ca2+ channel molecule,210,213 minor genetic variabilities of which can be connected to inherited chronic venous disease.75,79 Endothelial shear activates the Tie2 tyrosine kinase receptors in endothelial cells. 213 Flow-induced control of the morphological lumen could be proven in veins, even under the conditions of increased gravitational load.36,37,135
Human venous endothelium subjected to gravitational stress has secretory granules closer to the structure of Weibel–Palade bodies. 156 Disturbed endothelium releases endothelin, one of the most powerful vasoconstrictor agents. 214
Substantial functional and morphological endothelial damage can be observed both in early and later phases of the human varicosity disease.215–217 Seriously deformed varicose vein segments can be fully devoid of endothelium in large patches. 217 Endothelial samples from insufficient and varicose leg veins expressed more contact proteins, promoting the rolling, marginalizatio,n and finally the migration of white blood cells, at least in a certain phase of the disease.218–220 The intimal layer undergoes a substantial remodeling in varicose veins, and the dysfunctional endothelium plays a pivotal role in perpetuating the inflammatory cascade. 220
It is still questionable, how varicosity affects apoptotic processes in the venous endothelium. Apoptotic morphologic features, such as fuzzy mitochondrial cristae, lysosomal changes, and margination of the nuclear chromatin were found in varicose veins; still, the number of apoptotic cells was not higher than in healthy segments. A dysregulation rather than an explicit elevation of apoptosis seems probable.221,222 In varicose segments, there is a subintimal thickening with increased expression of the extracellular anchor protein laminin and reduced amount of COL type IV in the basement membrane. 215 Under the effects of venous hypertension endothelial luminal surface protein expression will be altered promoting the marginalization and migration of white blood cells into the venous wall. 219 Substantial involvement of endothelial cells in the pathological process of varicosity has been proven by Tisato. 220 She found that a substantial part of endothelial cells from varicose samples (C2 and C3 stages) were in the proinflammatory, migratory phase, with enhanced expressions of the endothelial adhesion molecule marked with the code number CD146, the neutrophil-binding CD31/PECAM-1 (platelet and cell adhesion molecule 1) and ICAM-1 (intercellular adhesion molecule; see also the “White Blood Cells, Inflammation and Metabolism” chapter). They expressed more cytoplasmic NF-κB (a transcription factor resetting cell functions to chronic inflammation), released more OPG (osteoprotegerin, with a function related to white-cell contact), and vascular endothelial growth factor (VEGF). Increased venous pressure increases the permeability of the endothelial layer. 223
A recent publication applied the most advanced single-cell mRNA expression survey technique to human saphenous vein specimens. A marked heterogeneity of endothelial cell populations could be proven. Significant differences have been found in the expression of atypical chemokine receptor subtypes. Such receptors are thought to bind chemokines without eliciting the activation cascade action. Of such receptors, the cell surface atypical chemokine receptor type 1 (ACKR1) demarcated nonvalvular endothelial cells, whereas other subtypes, ACRK3/ACKR4 were exclusively expressed by valvular endothelial cells. Such differences raise hopes to construct drugs selectively acting on specific types of venous endothelium. 224 Electronmicroscopic observations revealed endothelial cells with irregular borders, nucleus margination, cytoplasmic vacuoles, and degenerative signs on mitochondria.119,217,225 Flow cytometric study of endothelial cells harvested from different points of the varicose saphenous vein confirmed the inhomogeneous and dynamic character of the disease with much less expressions of E-selectin, VCAM-1 (vascular cell adhesion molecule 1), tissue factor (TF), and thrombospondin receptor(s) in the proximal segments than in the distal ones. 226
Smooth Muscle and Contractility
Smooth muscle contraction is the only possibility to shrink the lumen of a vessel following passive dilation under pressure (not to mention the very ineffective and slow intimal thickening process). Leg venous smooth muscle receives substantial sympathetic stimuli from the adventitial side, vasoactive stimuli from blood and from the surrounding tissues, and contains several vasoconstrictor and vasodilator receptors, even mechanical sensors that control its contraction state in the healthy wall.152,227–229
There are substantial changes in vascular smooth muscle during the development of the disease. We have to stress the characteristic heterogeneity of this disease: at the same time point, in the same network we will find areas with slight changes and also areas with advanced pathology. Even the pathological processes themselves demonstrate high variability: areas of cellular proliferation can alternate with those of cell degeneration, fibrotic transition, apoptosis, and even necrosis in the imminent neighborhood.230,231 We are convinced that by synthesizing published data now we are in the position to describe the sequence of events of pathological changes in the wall.40,232
All vessels, veins included are in the process of a slow, continuous remodeling of the wall.36,135,137–139,233,234 Even branching angles are optimized.
235
Such processes will slowly adjust the morphological lumen size to match flow, wall thickness, and composition to the pressure load. Pathological levels of hemodynamic load initiate the leg varicosity disease.33,236 Hemodynamic effects can reach the medial smooth muscle cells by three ways: flow-induced endothelial shear, stretch-induced endothelial damage, and increased wall stress sensed by the smooth muscle cells themselves. (1) Endothelial cells (as shown in the previous section) can sense shear force through flow in the lumen. The immediate effect is release of NO and acute endothelial vasodilation, which is present in veins
207
and venules.
134
Endothelial dilation is maintained in moderately affected varicose areas.
237
In addition to this acute effect, by pathways still not fully understood, chronic alterations in endothelial shear will induce the morphological remodeling of the lumen. At bifurcations (confluences), shear levels will be kept if the cube of the diameter of the mother branch is equal with the sums of the cubes of the diameters of daughter branches. We successfully proved that this so called “Murray’s law”
234
can be demonstrated in the popliteal venous confluence of the rat at normal and even at artificially elevated (gravitation) leg venous pressures.
135
Flow-induced remodeling means that force-bearing structures need to be degraded and under conditions of continuously acting stress, rebuilt to fit the new morphological lumen. We demonstrated the presence of increased mitotic activity, monocytic infiltration, and presence of smooth muscle cells with limited level of smooth muscle actin expression in the small collateral veins that were undergoing the process of flow-induced wall remodeling.
37
We are convinced that a large part of the “venous vessel wall weakness,” a frequent term used in early publications refers to veins in which extra flow initiated the flow-induced remodeling processes, transiently weakening the wall. (2) The previous subsection revealed that pathological level of stretch on endothelial cells induces significant changes in their phenotype. The endothelial layer will be permeable permitting transport of macromolecules from blood into the wall of the vessel. Alterations in the expression of adhesion proteins will induce marginalization and migration of white blood cells into the wall. Cytokines released by them will stimulate the transformation of contractile smooth muscle cells into the secretory, migratory phase,39,230,238 finally they stimulate apoptosis.221,231,239 Endothelial lesions will stimulate thrombocyte aggregation, and released substances penetrate into the wall with massive pathological consequences.
240
(3) Vascular smooth muscle cells have the ability to sense circumferential wall stress. The sensory molecule has been identified as a transient receptor potential molecule of type 6 (TRPC6), but the role of the angiotensin type 1 receptor (ATR1), mechanosensitive potassium channels, and of certain cell-surface integrin molecules has also been suspected.
129
Another transient receptor potential molecule (TRPM7) is able to initiate the transition of adventitial fibroblasts into smooth muscle actin containing myofibroblasts upon stretching through the p38 MAPK/Jnk (p38 mitogen-activated protein kinase/c-Jun N-terminal kinase) pathway.
241
Chronically increased wall stress within the physiological limits stimulates smooth muscle cell division, wall thickening with more developed muscular layers.137–139,155 Prolonged gravitation stress in the rat saphenous vein induced lumen dilation, elevation in wall mass, and hyperpolarization of vascular smooth muscle cells.
151
In the case of pathologic overstretch, cells will assume the secretory-migratory phenotype resulting subintimal cell accumulation and increased level of protocollagen expression. Damaged cells will leave the cell cycle and turn apoptotic in high numbers.221,231,239
Incompetent veins with no clinical symptoms contract normally, 237 with the appearance of varicose deformation, contractile ability will be lost.181,207,216,232,237,242–246 Smooth muscle cells from varicose segments released much less Ca2+ from vesicles when stimulated with agonists. 247 Smooth muscle cells produce contractile proteins, collagen, and elastin in the proper ratio. In response to overstretch of the wall and to inflammatory stimuli, smooth muscle cells will be transformed from the contractile to the secretory–migratory phase.39,230,238,248,249 Instead of the contractile proteins, protocollagen and adhesion proteins will be expressed. In varicose segments, disorganization of smooth muscle bundles is present, these are degenerated, vacuolized smooth muscle cells, and later during the process, fibrosis replaces the muscle cells.215,250 Apoptosis is frequent, appearing inhomogeneously and in the later phases of the pathological development. 231 An interesting observation is that smooth muscle cells in the vicinity of valves demonstrate higher rates of migration and proliferation. 251
Several growth factors and cytokines have been proven to be present in high quantities in varicose venous tissue, their identification raises hopes for a potential future pharmacological intervention. Increased contents of aFGF (acidic fibroblast growth factor), IGF-1 (insulin-like growth factor 1), and VEGF-A (vascular endothelial growth factor-A) and increased expression of the receptors IGF-1R (insulin-like growth factor 1 receptor), VEGF-R2 (vascular endothelial growth factor receptor type 2), and TGF-beta RII (transforming growth factor beta receptor type 2) have been found. 252 In a recent work, increased expression of the insulin receptor substrate 4 (IRS-4) could be established. It is stimulated by IGF-1 and had PI3K (phosphatidyl inositol 3 kinase) and MAPK (mitogen-activated protein kinase) downstream effectors. 253
While RANTES (regulated upon activation, normal T cell expressed and secreted, a chemotactic molecule for leukocytes), IP-10 (interleukin 10, interferon inducible protein), MIP1-β, and MIP1-α (macrophage inflammatory proteins) chemokine mRNAs were not expressed in intact veins, they were expressed in high amounts in varicose specimens. Further, MCP1 (monocyte chemoattractant protein 1) and IL-8 (interleukin 8) expressions were elevated. From these substances, IL-8 and MCP-1 are thought to be produced by fibroblasts and smooth muscle cells, the others by invading leukocytes. 254 TGFβ1 (transforming growth factor beta1) has been found to be expressed in the pathologic smooth muscle cells themselves. 230 This way smooth muscle cells are not only targets of the inflammatory processes initiated by venous hypertension and initial endothelium damage but active organizers of the inflammatory processes themselves. In transplanted grafts, a cross-talk between smooth muscle cells and monocytes was found. Monocyte IL-1β/IL-18 (interleukins) activated graft smooth muscle cells through the PI3K/AKT (phosphatidylinositol 3 kinase, Rho family serine-threonine kinase), mTOR (mammalian target of rapamycin), ERK1/2 (extracellular signal-regulated kinase ½), and STAT3 (signal transducer and activator of transcription 3) pathways. They induced expression of PDGF-bb, (platelet-derived growth factor-bb) one of the most effective stimulants of proliferation and migration of vascular smooth muscle cells, inducing thickening of the pressurized graft. 255 Phenotypic transition is marked by the reduced expression of contractile markers, such as smooth muscle 22[alpha], calponin, smooth muscle actin, and myosin heavy chain 11 in muscle cells prepared from varicose veins. 98 The forkhead box C2 (FOXC2) transcription factor, seems to be one regulator gene of smooth muscle transition from the contractile to the secretory–migratory phase, activated, amongst others by TGFβ. A recent observation has proven that a non-coding RNA fragment, the level of which is downregulated in varicose veins, microRNA (miR)-199a-5p has a target site on FOXC2, inhibiting it. 98 In animal experiments, we have observed that in a newly formed collateral venous network, in small veins being in the process of flow-induced wall remodeling, there was increased expression of the Ki67 (nuclear cell proliferation marker) protein, marking cell division activity and areas with insufficient expression of the contractile protein smooth muscle actin (SMA). As a result, when subjected to chronic gravitational load these veins were unable to resist elevated pressure and formed typical varicose malformations. 37
Venous hypertension has nuclear effects. In patients with venous reflux, there was a higher expression of the mRNA for PARP (poly ADP ribose polymerase, enzymes with central role in DNA reparation) in the saphenous vein walls even in young patients. 96
Final fibrosclerosis seems to be preceded by a proliferative phase of smooth muscle cells, but these smooth muscle cells will be eliminated by apoptosis. 239 Varicosity destroys the smooth muscle component of the venous wall in an inhomogeneous manner, the process will be more and less expressed at different places, and this prepares the soil for inhomogeneous dilations that cannot be controlled any further.232,237,256
Disintegration of smooth muscle layers seems to be the most important destructive event in the pathologic remodeling of the varicose vein wall.250,257 In a damaged vessel, the amount of smooth muscle component will be reduced and replaced with collagen. This change is histologically irreversible. While there is more and more collagen produced, and that will prevent vessel rupture (which could be fatal), slow creep of circumference (e.g., just a few micrometers per day) driven by high transmural pressure will result in a morphologically more and more dilated vessel. An analog mechanism was demonstrated in our lab for irreversible dilation of cerebral berry aneurysms. 258 This can be prevented only by surgical removal or closure (sclerotization, ablation) of the affected segment.
The most advanced single-cell RNA sequencing technique has recently been successfully applied to cells prepared from nonvaricose human saphenous veins. It revealed a substantial heterogeneity of the smooth muscle cell population. 224 Investigating and understanding differences between smooth muscle cell populations in varicose lesions seems to be a very promising area in the future.
Extracellular Matrix
The classical view considered venous varicosity to be a connective tissue pathology,120,259,260 not without reason. Most striking histological changes in the wall of an (advanced) varicose segment affect the connective tissue component. Veins subjected to high pressures will have, following an initial elevation of elastic tissue, fragmented elastic membranes and then the elastin will be diminished, and finally, it can fully disappear from the wall. Elastin and the smooth muscle component in the media will be replaced by rough bundles of collagen.185,260–264 Increased laminin expression will be found even in the subintimal space,215,262 while collagen type IV diminishes. 217 Fibrosis is driven by chronic inflammatory processes; collagens type I and type III will be expressed in an unbalanced manner. 265 Elastin to collagen ratio seems to be the most reliable factor when evaluating mechanical viability of a varicose segment. 266
Study of the distribution of collagens type I, II, and IV in different layers of the wall did not yield conclusive results. 257 Incompetent saphenous veins contained more type I collagen and less type IV collagen than competent ones. Interestingly, in this study the amount of elastin in the wall was also elevated, which can be explained by the fact that specimens collected for vascular reconstruction surgery were studied, with all probability segments carefully selected in the initial phase of the disease.
Increased venous pressure can elevate the expression of several MMPs (matrix metalloproteinases) through inflammation or tissue hypoxia and HIF (hypoxia inducible factor) expression. 267 Matrix metalloproteinase types 1, 2 and 9 were found to be elevated. 268 Jacob et al. 269 explained the accumulation of extracellular matrix in varicose veins by the observed elevation of the ratio of matrix metalloproteinase tissue inhibitors (TIMP-1) and the expressed matrix metalloproteinases (mostly MMP2). Connective tissue changes are slow to develop (theoretically allowing time for preventive and therapeutic interventions). During varicose remodeling, the expression of the whole set of enzymes governing connective tissue degradation and rebuilding are altered. 270
In the final stage of the disease, predominance of fibrous tissue, disorganization of the elastin/collagen lattice, and abnormal distribution of dystrophic elastic tissue are characteristic histological findings. 262 Increased amount of collagen type IV, laminin, and tenascin was found by Kirsch in varicose specimens. 120 On this basis, he theorized that pathology of the venous wall itself should be the primary cause of the disease, not the hemodynamic changes.
Synthesis of tropoelastin and fibrillin, essential components of elastic fibers is downregulated in certain areas of the media, where organized elastic fibers are missing. 271 Destruction of the elastic membranes causes the typical rigidity of the wall. 272 In newly formed collateral veins, the expression of elastica cannot keep up with the rate of the flow induced remodeling of the wall, this can explain their increased sensitivity to the gravitation load observed. 37 In the rat saphenous vein main branch with experimentally reduced flow, in the process of low flow-high pressure remodeling the lumen and the amount of wall material were reduced. During that wall-rebuilding process, density of the inner elastic membrane decreased, and new scattered elastic elements appeared in the media. 36 In experimentally produced, newly formed reticular veins a decreased amount of collagen was observed. 38 In varicose veins the association between smooth muscle cells and elastin was found to be disturbed. 273 In the varicose vein wall areas with normal elastin content and those with diminished elastin alternate, dilation of the wall seems to be associated with locally reduced elastin content. 264
Medial connective tissue is produced by vascular smooth muscle cells. Smooth muscle cells obtained from varicose specimens produced less fibronectin and collagen type III than normal cells, the difference seemed to be more translational than transcriptional. 274 Such cells expressed more collagen type I than normal ones; the difference was explained by the elevated transcriptional rate of mRNA. An imbalance between collagen types I and III (in favor of type I) was the result. 265 This imbalance was further enhanced by increased degradation of collagen type III by the MMP-3. 275
Even relatively young patients (<50 years old) with clinically established venous reflux had elevated COL I, COL III (Collagen type I and III) and matrix metalloproteinase MMP2 expressions, reduced inhibitor, TIMP 2 expression and higher COLI/COLIII ratios. These changes were attributed to DNA damage. 96 There seems to be a distinction between the mechanical functions of collagen types I and III, this latter characteristically is present in the wall of hollow organs subjected to substantial distension during their physiological function. 276 Venous segments subjected to mechanical overstretch expressed more metalloproteinases MMP-2 and MMP-9 under in vitro conditions. 181 There are substantial differences in the expression of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in different areas of the varicose network and in different phases of the disease, atrophic and hypertrophic areas showing large contrasts in this respect. To reveal the significance of these local differences requires further investigations. 277
Proteomic analysis confirmed the presence of >150 extracellular proteins expressed in the venous wall. Comparison of varicose saphenous vein segments with normal specimens revealed the loss of aggrecan and several small leucine-rich proteoglycans, they were replaced by increased amounts of type I collagen and laminins. Small leucine rich proteoglycans of the extracellular matrix (like decorin and others) have a horse-shoe shape and embrace collagen fibers shaping the diameter of the bundles. Chymase and tryptase β1 degradation enzymes were upregulated. The former degrades basement membranes, while the latter is very effective in the degradation of the extracellular matrix. 278 Fibronectin is another extracellular matrix protein with high degradation rates in the varicose vein wall. 279
White Blood Cells, Inflammation, and Metabolism
Inflammatory changes accompany lower extremity venous hypertension, varicosity, and venous insufficiency.131,206,280–284
Long standing venous hypertension induces inflammatory changes in the vein wall.37,206,218,285 Mechanical stress, especially extreme shear induced by local turbulence, induces transformation of endothelial cells which will be then more permeable. 223 Adhesion proteins on their luminal surface will be expressed promoting marginalization and migration of white blood cells into the wall. 45,182,218–220,281,286,287 These infiltrating cells are macrophage-monocytes, T lymphocytes, and even mastocytes. 286 Smooth muscle cells themselves are sensitive to mechanical overstretch induced by sustained high venous pressure and will be transformed into the secretory–migratory phase.129,248 White blood cells migrating into the wall release a series of inflammatory cytokines, which further amplify the transition of vascular smooth muscle cells into the secretory–migratory phase.182,239,254,288 Most of the proteases degrading the original connective tissue scaffold are produced by these migrating leukocytes, but smooth muscle cells also contribute to it. 268 Well-structured connective tissue will be replaced by disorganized clusters of collagen fibers produced by the transformed medial smooth muscle cells and adventitial fibroblasts. The typical histological picture of chronic inflammation with irreversible fibrosis emerges.185,260–264 A substantial part of varicose wall damage can be explained by these inflammatory effects. 230
Endothelial cells assume the proinflammatory phenotype in varicose segments, expressing the proinflammatory markers code-named CD31, CD168, as well as ICAM-1 (intercellular adhesion protein 1) and OPG (OsteProteGerin). 220
The requirement for morphological flow remodeling may further add to the inflammatory component. Macrophages infiltrate the venous wall and its surroundings during the process of flow remodeling to remove unfit for the new morphological lumen connective tissue sheets and perivascular components.36,37 A cross-talk between monocytes and smooth muscle cells is initiated through interleukin IL-1β/IL-18 induced release of PDGF-bb (platelet-derived growth factor-bb), a potent mitogen and chemoattractant for vascular smooth muscle cells. This process has been proven to play an important role in the thickening of the wall of venous grafts implanted into the arterial system. 255
Among the infiltrating inflammatory cells mast cells could be identified.286,287,289
A substantial part of tissue damage will be induced by these cells migrating into the wall. In addition to their cytokine and protease production, they produce reactive oxygen species. The significance of ROS (reactive oxygen species) in wall damage is shown by the fact that severity of the pathological process has been found to be inversely proportional to the anti-oxidative capacity and superoxide dismutase activity of the tissue. There is substantial lipid peroxidation damage to cells during varicose development. 290
In varicose veins, an upregulated expression of chemokine mRNAs for the macrophage chemoattractant protein-1 (MCP-1), interleukins IL-8, IP-10, the T cell chemoattractant (RANTES), and macrophage inflammatory proteins (MIP-1α and MIP-1β) were found. Their source was not the endothelium. 254
Lymphocytes removed from refluxing great saphenous veins released more cytokines, chemokines and growth factors when stimulated in vitro, the authors attribute the difference to the intermittent (turbulent?) flow in the deformed venous system. Upregulated cytokines were interleukins (IL-2, IL-4, and IL-12 p70) and interferon (IFN-γ); surprisingly, stimulated lymphocytes produced less tumor necrosis factor (TNF-α). Regarding chemokines and growth factors, release of macrophage inflammatory proteins (MIP-1A and MIP-1B), eosinophil chemotactic chemokine (eotaxin), and granulocyte colony stimulating factor (G-CSF) were found to be elevated.288,291 Blood itself, collected from varices contained elevated amount of IL-6. 284 Further, IL-12 and VEGF levels were elevated in the brachial vein blood even in early varicosities further demonstrating the inflammatory character of the disease. 292
The transforming growth factor (TGF-β1) seems to be a central extracellular signal in varicose remodeling of the venous wall.277,293–296 It stimulated the expression of matrix metalloproteinases and their inhibitors, MMP9, MMP12, TIMP1, and TIMP2. 295 Furthermore, it has been shown, that not only this agonist, but also its receptor, TGF-β RII, and its key intracellular signalization protein, in activated phosphorylated form (p-Smad2/3, only abbreviation is used) are also upregulated. 297 TGFβ expression is especially elevated in the final phase of the disease, by all probability connected to the intensive fibrosis. 294
More PDGF-BB was released from cultured endothelial cells obtained from varicose specimens. Its release was stimulated by exogenous TNF-α through the NF-κB pathway. 298 NF-κB is a central intracellular signal protein of chronic inflammation, common with TGFβ pathway; however, no data on PDGF-BB release in response to TGFβ could be found.
Somewhat contradictory observations were made by Gomez 299 who failed to demonstrate the presence of inflammatory cells or elevated levels of any of the common inflammatory markers, the C reactive protein (CRP), fibrinogen, PTX-3 (pentraxin-related protein), the secretory phospholipase sPLA2-IIA, and cyclooxygenase type 2 enzyme (COX-2) in saphenous vein samples, declared to be in the C2 phase of the disease. The explanation again, we believe, is that these specimens were collected for coronary bypass surgery, and we have a good reason to think that minimally affected segments have been carefully selected and harvested. Anyway, this draws our attention again to the heterogeneity of the pathology of the disease.
In a newer experimental study, several important steps of the finer molecular mechanisms of the high pressure induced pathological venous wall remodeling have been identified. Murine venous segments have been subjected to high arterial pressure being inserted as bypasses. They released the proinflammatory cytokines IL-1β/IL-18 during the process of their arterialization. In vitro venous tissue debris activated monocytes to release these same substances. Cultured venous smooth muscle cells were then activated through the conventional PI3K/AKT, MAPK and STAT3 pathways by these activated monocytes as well as by the cytokines released by them. Even the release of the potent mitogen and chemoattractant of vascular smooth muscle cells, PDGF-bb was stimulated. 255 Interleukin IL-17 was found in elevated amounts in insufficient young veins by Ortega. 96
Inflammatory components amplify mechanical overburden-induced apoptotic tendency of vascular smooth muscle cells. As we mentioned earlier, dysregulation with patchy areas of normal and elevated apoptotic processes, rather than an explicit elevation of apoptosis appears to be probable.221,222
A recent observation added important elements to our knowledge on the inflammatory pathways during the development of the varicose vein wall. Upregulation of more than 200 circRNAs has been observed, several of them connected to the NF-κB (chronic inflammation) pathway processes. 100
An interesting question is what metabolic processes can play a role in the pathomechanism of varicosity in addition to the explicit inflammatory and apoptotic reactions discussed above. Tissue flow deficit and local hypoxia are the most important factors in the development of the most serious consequences: venous ulcers (see the corresponding section). Stasis, depleting blood oxygen, and wall thickening interfering with oxygen diffusion can induce hypoxia and cell damage in the venous wall itself. 256 Again, this will be characteristic in the most advanced forms of the disease, the significance of which is frequently underestimated. 300 Endothelial cells are sensitive to hypoxia. 301 Smooth muscle cells from insufficient venous walls expressed more mRNA of the HIF-1α protein (hypoxia inducible factor 1 alpha, an intracellular regulator of resetting cell metabolism to hypoxic conditions), both in normoxic and hypoxic culture conditions. 302 HIF-1α protein is responsible for the increased VEGF production in tissues and in the vasculature, the saphenous vein wall included. The CLOCK gene product (circadian control, DNA nucleotide excision repair, apoptosis checkpoint control) seems to stimulate HIF-1α production especially in advanced forms of varicosity. 303 Increased pressures induce thickening of the wall, which can be accompanied by hypoxia and compensatory development of vasa vasorum from the adventitial side.302,304
Platelets and Thrombosis
Intact endothelium and regular laminar flow prevent the activation of clotting factors and adhesion of platelets.203,205,206,305 These characteristics are massively disturbed at the site of varicosities. 217 Disturbed endothelium produces instead of the antiaggregatory agonists NO134,207 and prostacyclin, 208 endothelin 214 with proaggregatory actions.306,307 Cell apoptosis seems to be connected to manifest superficial vein thrombosis. 308 Thrombophlebitis further aggravates the inflammatory remodeling of the venous wall. 309 It appears that platelets play an important role in inducing the morphological deformation of the varicose venous wall. Contribution of platelet derived growth factor (PDGF) to vascular aging, intimal and medial thickening, vascular stiffness have been reviewed by Yang. 240 Endothelial damage at sites of turbulent flow can stimulate local thrombocyte aggregation, released PDGF further deforms lumen contour, forming thus an irreversible vicious circle.
There seems to be a connection between the blood clotting system and the venous wall proneness for chronic venous disease. In a meta-analysis, Tan 191 found a connection between thrombophilic genetic disorders and the occurrence of non-thrombotic venous disease. Two earlier publications from our lab had proven that the in vivo rigidity of the common femoral vein increases both in non-thrombotic thrombophilic patients and also in the post-thrombotic state even at the non-affected side.192,310
In the early phase of the varicosity disease, in blood samples taken from the brachial vein there were no changes in fibrinogen, protein C and tissue plasminogen activator (tPA) concentrations, however, amount of protein S and of the von Willebrand factor (vWF) were elevated. 292 The von Willebrand factor (vWF) is released from activated endothelial cells, and in addition to its main function serving as a molecular bridge to attach platelets to collagen, it also interacts with leukocytes and can promote inflammatory reactions. 311 We found that secretory granules in the endothelial cells of the human leg small veins (healthy) were more elongated, and their structure was closer to the Weibel–Palade bodies (known to contain vWF) than those of small veins of the neck. This proves a direct connection between venous hypertension and local thrombotic processes in humans whose lower extremity is exposed to high pressures because of bipedalism. 156
Interaction of cellular processes during the initiation and development of the chronic venous disease of the lower extremity is shown in Figure 2. One can identify numerous (several hundreds) potential positive-feedback circuits. Pathomechanism of lower extremity chronic venous disease. Cellular and molecular mechanisms. Pathological events for different cell/tissue types of the venous wall are organized in vertical columns, in logical order from above to the bottom. Bottom row(s): Developed cell/tissue pathology causing hemodynamic effects. First column, hemodynamic effects. Note numerous potential positive feed-back circuits. For a more detailed analysis of hemodynamics, see Figure 1.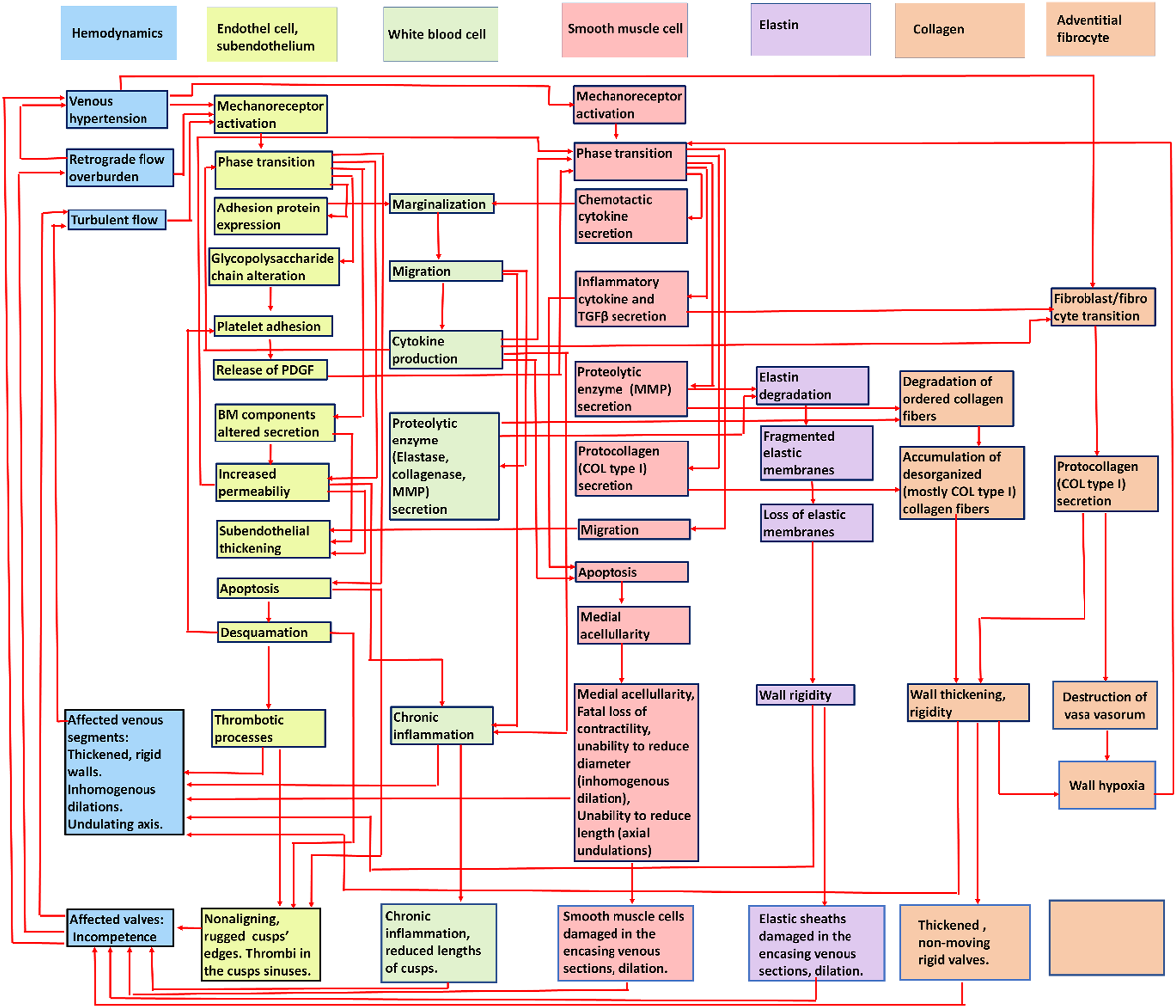
Endocrine and Neural Control
Both competent and incompetent superficial leg veins dilate during pregnancy, there is a recovery in the postpartum period. 312 Number of pregnancies does not seem to influence the reflux pattern. 313 However, the exacerbation of varicosity during pregnancy is well known.6,8,15,48,50 It is not clear to what degree it can be attributed to pelvic venous flow disturbance, weight gain or the high levels of female sex hormones. Preexisting varicosity reflux exacerbates during pregnancy, with improvement after delivery, but not in case of every refluxing vein. 314 Ovariectomy reduced agonist-induced contractility of the rat saphenous vein, and it was restored after hormone replacement therapy. 315 Saphenous veins of female rats in which a polycystic ovary disease was induced by androgen treatment had thicker walls and reduced contractility, reduced endothelial dilation. This disturbance was partially restored by vitamin D treatment. 316 A connection between varicosity and female sex hormones has been clearly shown by Serra 317 who found a substantial elevation in the expression of estrogen receptors (Erα, Erβ, and the G protein coupled estradiol membrane receptor, GPER) in the venous wall with advancement of the disease (increasing CEAP grades).
In patients with diabetes metformin reduces the risk of varicose veins. 318 Good glycemic control improves results after ablation in diabetics with better edema relief and faster ulcer healing. 319
The saphenous vein is richly innervated by postganglionic sympathetic fibers 320 and is an active participant of the orthostatic cardiovascular reflexes. Rats kept chronically in tilted cages to elevate gravitational load developed more sympathetic nerve terminals and more synaptic vesicles in the wall of the saphenous vein than control animals kept in traditional cages. 152 Taking into consideration that smooth muscle contraction is the only means to reduce distended vascular lumen, our knowledge on the sympathetic innervation of the side-branches of the saphenous vein, their alterations with the remodeling of the whole saphenous vein network during chronic venous disease seems to be unduly limited. Healthy and varicose saphenous veins, even main branches and side branches have different distribution of the alpha1 and alpha2 adrenergic receptors, raising hope for an effective, selective pharmacological intervention. 321
Morphological Disturbance in Superficial Network
A more detailed description of the clinical anatomy of low extremity varicose networks is possible with the application of the new techniques, such as contrast enhanced 3D CT venography, 322 advanced Duplex ultrasonography,323,324 intraluminal visualization during endoscopic surgery, 325 for superficial veins LED transillumination 29 and near infrared imaging.30–32
Telangiectasias are small, dilated veins in the skin (diameters 0.1-1.0 mm), reticular veins are forming outlined clusters of networking small veins (diameters 1-3 mm), if three or more small, dilated veins are branching away from a central point, the “spider vein” expression is used. Varicose veins are dilated, their lumen is uneven with local dilations, their course is typically tortuous, they have a deformed lumen shape, unevenly thickened walls. Diameter is usually >3 mm.121,244,323,325–329
Advanced ultrasonographic techniques reveal that even the earlier phases of the varicosity disease are connected to valvular incompetence. Santiago found that lateral thigh telangiectasias were associated with incompetent reticular and perforator veins. 324 Telangiectasias fed by reticular veins were found in the Edinburgh study in large numbers without observable trunk varicosity. 42
Varicose veins differ from aging veins. 35 The endothelium is partly missing, the endothelial cells are degenerative, and there is a subintimal thickening. 217 The media is mostly thickened, but thinned in certain areas, and in advanced cases, the smooth muscle cells are sparse or fully missing. 250 Elastica is fragmented or missing. Smooth muscle is replaced by irregular bundles of collagen.35,262
How do these characteristic macroscopic deformations develop? Telangiectasias and reticular veins are usually associated with incompetent perforators or feeder veins. 324 In rat experiments, we could produce typical reticular vein plaques and spider veins on the surface of the thigh muscles by chronically (13 weeks) narrowing the deep femoral vein. Such appeared at the surfacing point of an incompetent perforator vein.38 Small vein morphological dilation seemed to be connected to chronically elevated flow, the random position of valves along the venular network determined irregular flow pathways in the reverse direction. Extra volume loaded onto the saphenous tributary system induced varicose deformations there.37,38 Irregular bypassing veins developed when the main branch of the saphenous vein was narrowed. Certain optimal routes strengthened as the result of flow-induced lumen enlargement. In the process of flow remodeling, original connective tissue scaffold of the venous wall was destroyed, smooth muscle cells divided, but time was necessary to express sufficient amount of contractile proteins and new connective tissue sheets for altered circumference. Weakened wall was the result, and gravitational load produced varicose deformations not seen in veins not in the process of flow remodeling. 37
The underlying reason for the typical tortuosity can be axial buckling of the vessel embedded in a soft tissue, especially if elevated pressures do act following the destruction of the elastic elements.330,331 Uneven diameter can be the result of an inhomogeneous damage of the endothelium by elevated flow (potentially turbulent) and pressure. Platelets attaching to the wall, migrating leukocytes release substances (PDGF, cytokines etc.; see the corresponding sections above) influencing the remodeling of the media in an inhomogeneous manner. With inhomogeneous destruction of the smooth muscle cells the ability to reduce diameter will be lost in certain locations. This view is supported by observations by Labropoulos 323 who found that varicosities are more common in the tributaries of the great saphenous vein than in the main branch itself. It is the side branches, which are especially affected by flow overburden and turbulent flow from incompetent perforators.
Microcirculatory Disturbance and Ulcers
The most serious consequence of chronically elevated venous pressure is local necrosis of leg skin and underlying tissue: venous ulcers.118,144 For obvious reasons, capillary hydrostatic pressure is between arteriolar and venular pressures. Chronically high venous pressures elevate capillary filtration pressures and damage capillary endothelium, reduce capillary flow due to elevated pressures in the draining veins and by the elevated hydrostatic pressures in the interstitium compressing the microvessels.219,332 In serious venous insufficiency, avascular areas can appear, and at the base of ulcers, no visible capillaries are present. 333 Captured white blood cells and cell debris in the capillary further compromise local microcirculation.144,219,280,281,334 With microcirculatory insufficiency, hypoxic damage will affect tissues supplied by these vessels, mostly skin tissues, white blood cells will migrate not only into the vascular wall but into the supplied tissues, releasing inflammatory cytokines there. 219 The oxygen demand of leg tissues at rest is not too high, and hypoxia of the whole lower leg will induce pain upon walking. 335 At certain sites, blood supply to skin and underlying tissues can be reduced below critical level, and local necrosis with ulcers can develop as a final phase of the disease.118,144,219,332 Fibrinogen exudate may form a fibrin cuff around the vessels further inhibiting the diffusion processes.336,337 Growth factors essential for wound healing can be trapped by fibrin. 333 Bacterial infection can aggravate the situation.146,337
Conclusion
A state of the art classical review on the pathomechanism of the lower extremity chronic venous disease has been provided by Bergan et al. in 2006. 236 They proposed disturbed hemodynamics as the leading cause of chronic venous disease and emphasized the importance of ensuing inflammatory processes in the wall. With a series of new publications at hand, that provide a more detailed understanding of hemodynamics as well as of molecular and cellular events in the vein wall, we can confirm their original view. However, now we are inclined to think that physiological remodeling is continuously ongoing in the venous wall and that early varicose remodeling is a derailment of such processes induced by limited adaptation ability determined by genetic factors. Genetic adaptation of humans to bipedalism is generally sufficient, but it is limited against lengthy periods of standing and sitting: relative new forms of workload in industrialized societies. Vicious circles emerge between the irregular local venous flow and pressure and pathological remodeling of the wall, with destruction of the tiny structures of venous valves, leading to irreversible, slowly progressive morphological and functional damage of the whole venous system of the lower extremity.
Such vicious circles should be interrupted by preventive regulations, conservative therapy and later by surgical and endovascular interventional means.
Footnotes
Acknowledgments
Venous research in the laboratory has been supported by Hungarian National grants OTKA TO 32019, OTKA 42670, and NVKP_16-1-2016-0004AF and by a grant from the Dean of the Medical Faculty, Semmelweis University. For valuable advice, we thank Professor Peter Gloviczki MD, Mayo Clinic, Rochester, Minnesota, USA, and Chief Doctor Imre Bihari MD, PhD, A + B Clinics, Budapest, Hungary.
Declaration Of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Hungarian Science Foundation (OTKA TO 32019, OTKA TO 42670, and NVKP_16-1-2016-0004A).