
Abstract
Background
Neuroblastomas are the most common extracranial solid malignancy in children with variable manifestations and complications depending on the presence of paraneoplastic syndromes.
Materials and Methods
We performed a single institution retrospective cohort study of all patients less than 18 years old diagnosed with neuroblastoma or ganglioneuroblastoma between January 2002 and July 2022. Patients were identified through the pathology and cancer registry and cross-referenced with pediatric records. Patient demographics, clinical presentation, treatment, and outcomes were collected. A univariate descriptive analysis of the collected data was conducted.
Results
In our study period, 130 children were diagnosed with neuroblastoma, and 15 were diagnosed with ganglioneuroblastoma. There were 12 children with a paraneoplastic syndrome identified, 8 with NBL and 4 with ganglioneuroblastoma (GNBL). The average age at diagnosis was 22 months. All but 1 underwent resection prior to treatment of paraneoplastic syndrome, and 4 children required neoadjuvant therapy. Neurological complications were the most common with 10 children (83%). The average time from symptom onset to diagnosis was 0.7 months. Eight children had complete resolution of their symptoms after treatment and resection, 2 children recently started treatment within a year, 1 had partial resolution, and 1 died during treatment. The presence of tumor-infiltrating lymphocytes occurred in 4 children with neurologic paraneoplastic syndromes. Six children had neuropil rich tumors.
Conclusion
The histological profile of paraneoplastic syndromes of neuroblastoma and ganglioneuroblastoma and their treatment across a single institution can be highly variable. The presence of tumor-infiltrating lymphocytes and neuropil may have an impact on paraneoplastic pathology.
Background
Neuroblastoma is the most common extracranial solid tumor of childhood, occurring at an average age just under 2 years. 1 Along with ganglioneuroblastomas and ganglioneuromas, neuroblastomas represent a spectrum of peripheral neuroblastic tumors arising from primitive neural crest cells of the sympathetic nervous system. 2 The clinical presentation and ultimate prognosis of these malignancies depends on several factors, including anatomic location, patient age, degree of local or distant spread, genomic alterations, and tissue pathology.
Histopathology after tumor resection or biopsy confirms the diagnosis of neuroblastic tumors and, coupled with molecular analysis, informs risk categorization and ultimately prognosis of disease. Histologically, neuroblastoma is characterized by small round blue cells with scant cytoplasm, known as neuroblasts. The International Neuroblastoma Pathology Classification (INPC) system was developed to characterize these neuroblastic tumors based on the degree of Schwannian stromal development (stroma-rich, stroma-dominant, stroma-poor), grade of tumor differentiation (differentiating, poorly differentiating, and undifferentiated), mitosis-karyorrhexis index (MKI) and patient age, ultimately defining the tumor histology as favorable or unfavorable. 3 Schwannian stroma is characterized by spindle-shaped fibroblasts admixed with Schwann cells, within a background of neuritic processes.
Neuroblastoma manifests in curiously protean ways. Interestingly, a subset of these patients presents with unique constellations of symptoms that manifest secondary to development of a paraneoplastic syndrome. Opsoclonus-myoclonus syndrome (OMS) – also known as Opsoclonus-Myoclonus-Ataxia Syndrome, OMA, or OMAS – is the most common paraneoplastic syndrome seen in patients with neuroblastic tumors. OMS is characterized by involuntary eye movements, muscle jerks, and ataxia. OMS is thought to be caused by the production of autoantibodies to tumor antigens that demonstrate cross-reactivity with normal neural tissue.1,4 Another that occurs less commonly in patients with neuroblastoma is characterized by profuse watery diarrhea secondary to overproduction of vasoactive intestinal peptide (VIP) secreted by the tumor. 5 The incidence of OMS-associated neuroblastoma at presentation has been estimated to be between 2% and 3%. 4 OMS has also been documented to arise on rare occasion during therapy, presumably as primitive neuroblastic cells differentiate. Cases of VIP-associated neuroblastoma are much less frequently documented in the literature. According to a retrospective study conducted by Bourdeaut et al, less than 1% of all neuroblastic tumors demonstrate clinical evidence of VIP secretion. 6 Even more rarely, a subset of patients with neuroblastoma present with elevated parathyroid hormone-related peptide (PTHrP), although the clinical presentations of these patients are not well-documented in the literature. 7
Given the heterogeneity in clinical presentation of neuroblastoma, several studies have sought to determine whether unique histologic features may retrospectively distinguish patients with neuroblastoma presenting with paraneoplastic syndrome. Two such studies by Cooper et al and Gambini et al focused on the histopathologic evaluation of neuroblastoma with associated OMS in a case-control series.8,9 They concluded that most cases of neuroblastoma presenting with OMS were ultimately classified as having favorable histology by INPC classification. Furthermore, they noted that the majority of cases had diffuse and extensive lymphocytic infiltration with lymphoid follicles, as compared to controls without OMS.8,9 Literature has shown that OMS-associated neuroblastic tumors generally have favorable oncologic outcomes and excellent response to treatment.10-12 Less is known about the histopathology of VIP-secreting tumors, likely as a result of decreased incidence in comparison to OMS. One study showed that VIP-secreting neuroblastomas demonstrated more differentiated phenotype with a marked predominance of stroma-rich or stroma-dominant tumors. 6 It is generally believed that VIP-secreting tumors, like OMS-associated tumors, also have more favorable outcomes and response to treatment. 4
In this study, we aimed to characterize neuroblastic tumor profiles in pediatric patients who experienced symptoms consistent with a known paraneoplastic syndrome. More specifically, we focus on histopathological features of the tumor and patient response to treatment. Our central hypothesis was that paraneoplastic symptoms aligned with a more differentiated histology.
Methods
This retrospective study received approval by the institutional review board of Vanderbilt University Medical Center (IRB #100734) and utilized a database of all children less than 18 years of age at diagnosis, who were treated for neuroblastoma and ganglioneuroblastoma as identified through the Vanderbilt Cancer Registry and department of pathology between January 1, 2002, and December 1, 2022 (n = 145). We then selected the patients in whom a paraneoplastic syndrome was identified in the electronic medical record (n = 12). Patients who underwent resection at an outside hospital were excluded, as detailed pathology reports were frequently unavailable.
Patient demographic and oncologic data was collected from the VCR and electronic medical record including age, race, gender, symptoms on presentation, age at diagnosis, stage (International Neuroblastoma Risk Group Staging System, INRGSS), Children’s Oncology Group (COG) risk stratification, neoadjuvant chemotherapy, operative details, treatment regimens, response to treatment at 1 year, and presence or absence of relapse. Paraneoplastic syndrome markers were also evaluated through the electronic medical record, such as PTHrP and calcium levels, and VIP levels. Additional oncologic and tumor histologic data such as tumor origin, grade, MYCN amplification status, and presence of lymphocytic infiltrates were derived from the final pathology report from the surgical specimen at the time of resection.
Statistical Analysis
Median and interquartile range were used to summarize data. A univariate descriptive analysis of the collected data was conducted. All data were analyzed using Microsoft Excel (Microsoft, Redmond, Washington)
Results
Cohort Description
During the study period, 130 patients were diagnosed with neuroblastoma (NBL), and 15 were diagnosed with ganglioneuroblastoma (GNBL). A paraneoplastic syndrome was identified in 12 children: 8 with NBL and 4 with GNBL. Ten children presented with OMS, while 1 child presented with profound watery diarrhea, concomitant electrolyte derangement, and elevated levels of VIP. The remaining patient presented with positive PTHrP. Tumor site was adrenal in origin for 9 patients (75%) and extra-adrenal for 3 patients (25%). Female to male ratio was 2:1, and the median age at diagnosis was 20 months (IQR 18.25, 28.75). All but 1 patient underwent resection prior to treatment of the paraneoplastic syndrome (Figure 1), and 4 patients required neoadjuvant therapy for non-low risk tumors. Of the 11 patients who underwent resection, 7 utilized minimally invasive technique while 4 were open resections. This case of VIP shows an emaciated child (A). CT (B) shows left adrenal NBL (asterisk). Note colon is full of fluid. (C,D) show the NBL at laparoscopy. Asterisk marks tumor. AV – adrenal vein.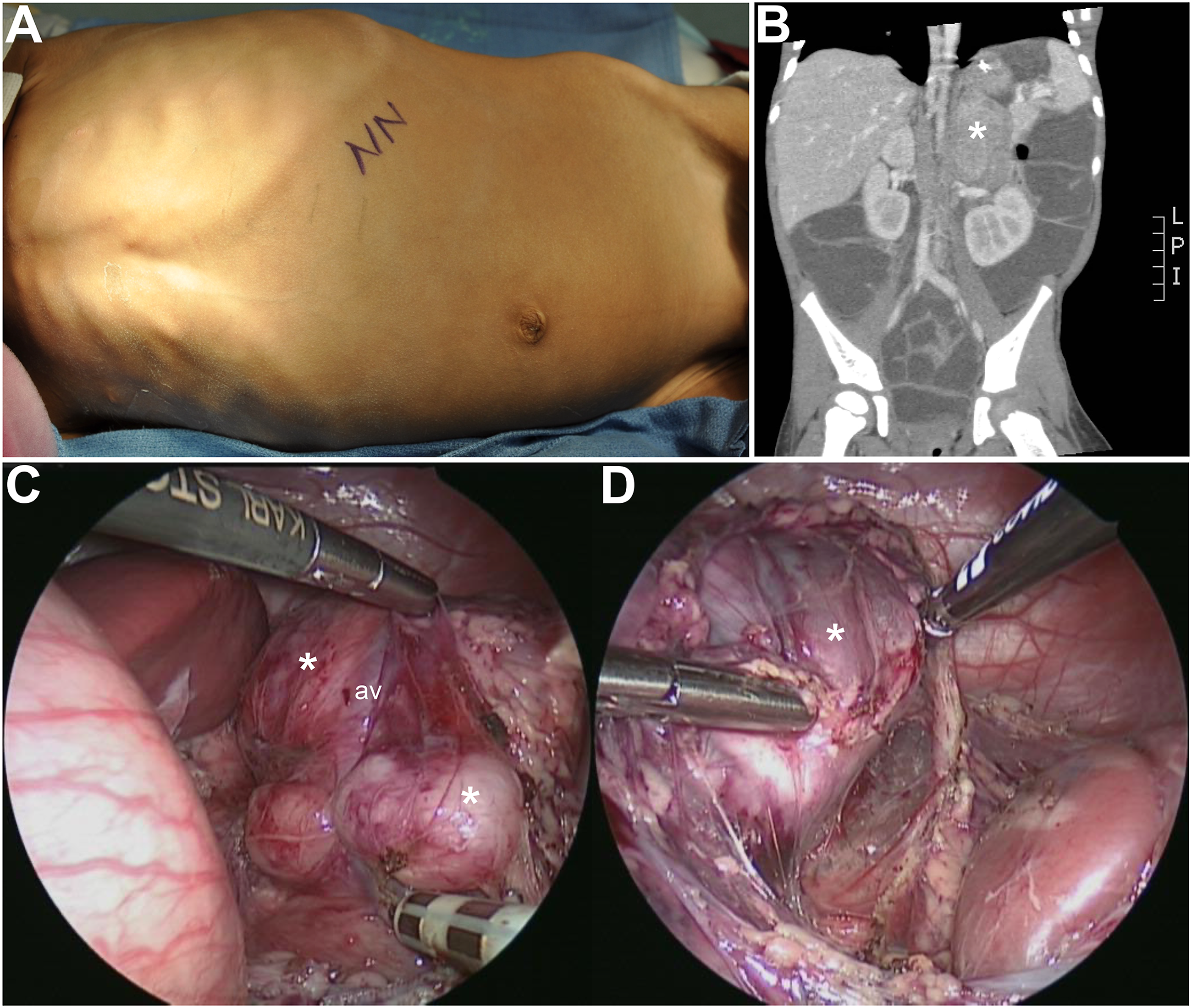
Paraneoplastic Outcomes
Paraneoplastic syndromes with neurological complications were the most common with 10 patients (83%). Eight patients demonstrated complete resolution of their symptoms following treatment. Two patients recently started treatment and have not yet reached 1 year to report symptom response. One patient had partial resolution and 1 patient died during treatment. The average time from symptom onset to diagnosis of neuroblastoma was .7 months. Patients who showed clinical improvement of symptoms related to paraneoplastic syndrome experienced resolution between 6 and 40 months of initiation of treatment, with a median of 15 months.
Histology
Patient Characteristics and Outcomes.
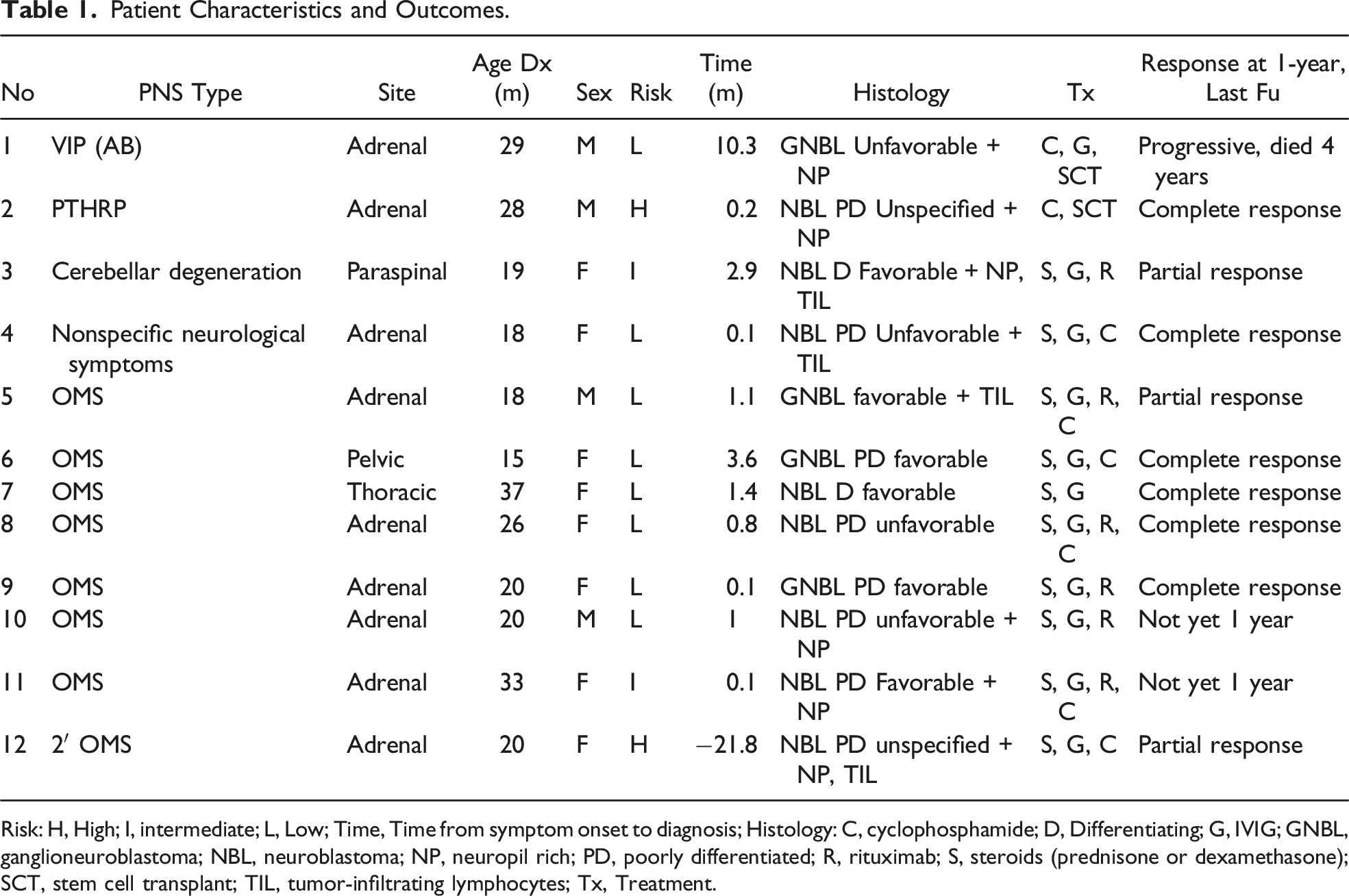
Risk: H, High; I, intermediate; L, Low; Time, Time from symptom onset to diagnosis; Histology: C, cyclophosphamide; D, Differentiating; G, IVIG; GNBL, ganglioneuroblastoma; NBL, neuroblastoma; NP, neuropil rich; PD, poorly differentiated; R, rituximab; S, steroids (prednisone or dexamethasone); SCT, stem cell transplant; TIL, tumor-infiltrating lymphocytes; Tx, Treatment.
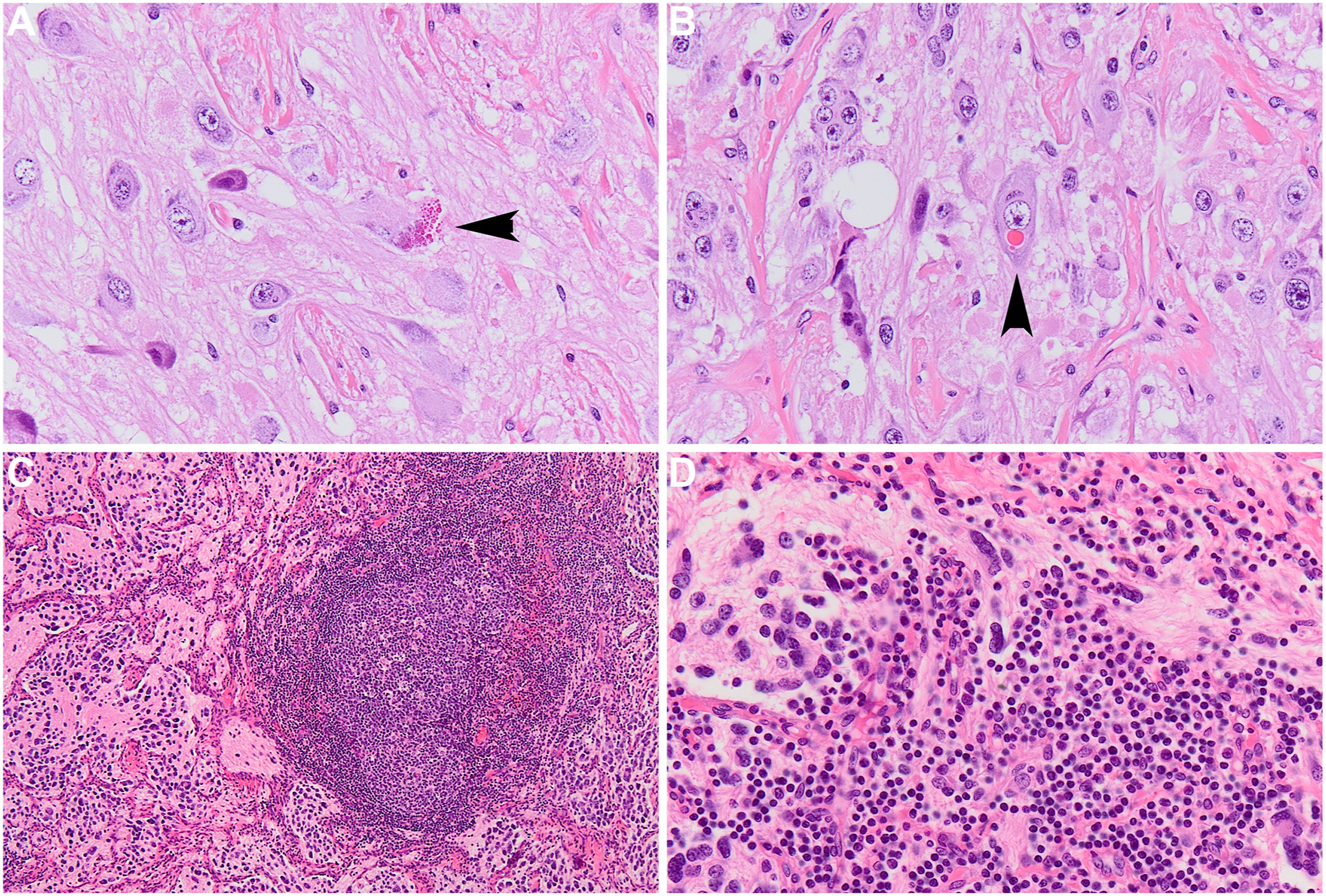
(A, B) Rare ganglion cells of a ganglioneuroblastoma with VIP. Arrowheads show cells with neurosecretory granules (hot pink vesicles in cytosol show prominent hyaline eosinophilic globules within their cytoplasm). Such globules are an unusual finding in neuroblastic lesions and may correspond to the VIP secretion identified in the clinical presentation. (C, D) This poorly differentiated neuroblastoma with OMS shows extensive lymphoid follicular hyperplasia throughout the lesion with the smallest cells depicting the lymphocytic infiltrates. A lymphoid follicle with germinal center formation is shown (C), set in a background of neuropil and neuroblasts. In areas (D), the lymphocytes are intimately involved with the neuroblastic elements, including the neuropil.
Discussion
Prior literature has demonstrated that OMS is likely a secondary immunologic event, following autoantigen recognition by lymphocytes surrounding or infiltrating differentiating regions of neuroblastoma. 13 These lymphocytic infiltrates, according to a study by Gambini et al, may be found within a perivascular, interstitial, or follicular pattern of formation. 9 Studies have sought to characterize autoantibodies from sera of patients with neuroblastoma and noted nonspecific reactivity to both neurons and cerebellar Purkinje cells. 13 However, no antigen candidate has been definitively implicated in the development of OMS-related autoantibodies. Thus, we sought to retrospectively review histologic features of patients with known OMS, seeking to find additional clues which could provide further insight regarding the development of this condition. Most notably, our findings suggest that OMS in patients with peripheral neuroblastic tumors may be a phenomenon more closely associated with tumors demonstrating some degree of stromal maturation, as defined by the INPC. 14 According to INPC guidelines, peripheral neuroblastic tumors may be categorized into 4 histologic subtypes: “neuroblastoma (Schwannian stroma-poor); ganglioneuroblastoma, intermixed (Schwannian stroma-rich); ganglioneuroma (Schwannian stroma-dominant); and ganglioneuroblastoma, nodular (composite: Schwannian stroma-rich/stroma-dominant and stroma-poor).” 14 Poorly differentiated neuroblastomas, although considered “Schwannian stroma-poor,” may be distinguished from undifferentiated neuroblastomas by the presence of background neuropil. Ganglioneuroblastomas and ganglioneuromas both demonstrate a predominance of Schwannian stroma. We noted that within our cohort of patients, none of the patients demonstrating OMS had the undifferentiated histologic subtype, which is the only histologic subtype among the peripheral neuroblastic tumors characterized neither by background neuropil nor Schwannian stroma. Given this observation, we hypothesize that the key antigen(s) recognized by lymphocytic infiltrates within the tumor and leading to the development of OMS may be unique to differentiating neuroblasts and/or differentiated neuroblasts with a ganglion cell phenotype. An alternative hypothesis could be that the key antigens reside within the background tumor neuropil or Schwannian stroma, which is not present in tumors of the undifferentiated histologic subtype. However, the latter hypothesis is less likely as Schwannian stroma predominance is preceded by the development of the intermediate and late stages of neuronal maturation.
The authors acknowledge several limitations with the study that temper rigorous conclusions. Foremost, the frequency of paraneoplastic syndromes is low and dominated by OMS when occurring. As a result, despite having a large experience treating neuroblastoma at our comprehensive pediatric hospital, the study power will be low and left to descriptive interpretation only. Further, the retrospective nature of the study relied upon pathology reports for the older cases, which were filed away at an off-campus site, complicating re-review of the actual histology slides. We also would preferred to have analyzed cell-surface markers for antigens to which the lymphocytes might be responding and to the content of the neurosecretory granules to determine whether the latter contained VIP.
In summary, this series attempted to address the histologic specifics of neuroblastic tumors associated with paraneoplastic syndromes. Indeed, no patient in our large, single institution series presenting with a paraneoplastic syndrome had an undifferentiated neuroblastoma.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.