
Abstract
A 27-year-old female presented at 13 weeks’ gestation with epigastric pain and anemia requiring blood and iron transfusions but no family history of gastrointestinal malignancy. Upper endoscopy revealed a giant circumferential polyp and associated hyperplastic-appearing polyps in the proximal stomach. Biopsies revealed hyperplasia with lamina propria eosinophils. She was supported with intermittent transfusions until labor was induced at 34 weeks’ gestation. Total gastrectomy was performed at seven weeks post-partum. Final pathology revealed multiple hamartomatous polyps without malignancy. Her anemia resolved postoperatively. Genetic testing revealed mutation of the SMAD4 gene and Juvenile Polyposis Syndrome. JPS is caused by germline mutations in the SMAD4 or BMPR1A genes and is characterized by hamartomatous polyps in the gastrointestinal tract. While most polyps are benign, malignant transformation can occur. One should have low threshold to send patients for genetic screening when multiple polyps are found in a young patient, even without family history.
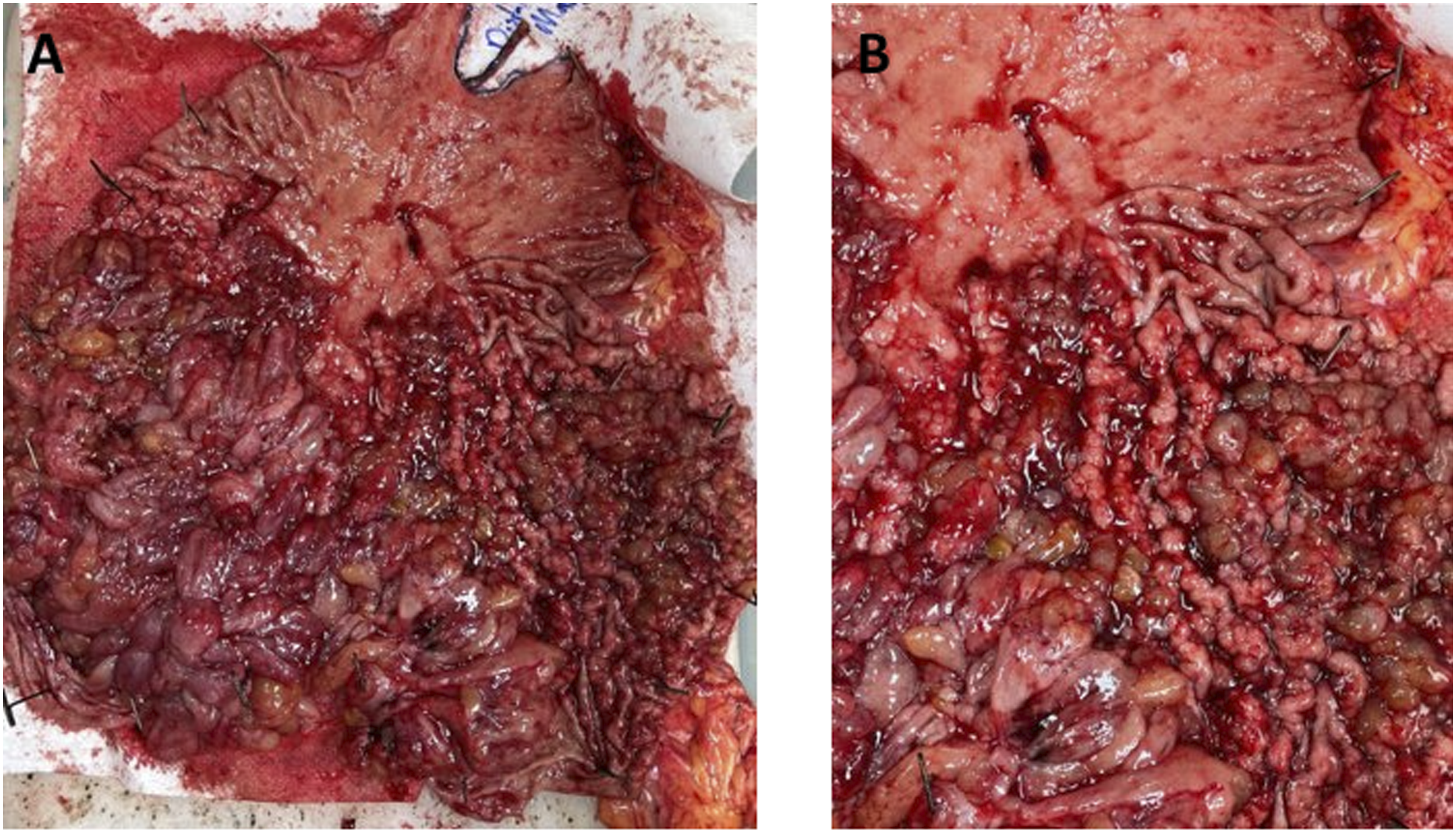
A 27-year-old female presented at 13 weeks’ gestation with recurrent epigastric pain and anemia. She had a several year history of iron deficiency anemia requiring blood and iron transfusions. She had no personal or family history of gastrointestinal neoplasia. Upper endoscopy revealed a giant circumferential polyp in the proximal stomach. Cross sectional imaging confirmed a 9.6 × 11.6 cm polypoid mass in the gastric fundus and proximal body without evidence of locoregional lymphadenopathy or metastatic tumors. Repeat endoscopy with endoscopic ultrasound evaluation revealed that the mass extensively involved the proximal stomach include the gastroesophageal junction. It appeared hyperplastic, and had a wide-based stalk. Additionally, there were hyperplastic changes diffusely throughout the rest of the stomach, distinct from the polypoid mass. Endosonographically, the mass was isoechoic, but without invasion into the muscularis propria. Biopsies revealed hyperplastic mucosa with frequent eosinophils in the lamina propria, without evidence of dysplasia or malignancy (Figure 1). Her care was coordinated between surgical oncology, high risk obstetrics, and her local hematologist. Due to persistent anemia and the concern for occult malignancy in the mass, she was supported with intermittent transfusions until 34 weeks’ gestation, and then labor was induced. After uneventful vaginal delivery of her son, total gastrectomy with pouch reconstruction and jejunostomy tube placement was performed at 7 weeks post-partum. Final pathology of the operative specimen revealed multiple hyperplastic/hamartomatous polyps, negative for dysplasia or invasive cancer (Figure 2). She had an uncomplicated postoperative recovery, with resolution of her anemia. After genetic counseling, germline testing revealed monoallelic mutation of the SMAD4 gene and a diagnosis of Juvenile Polyposis Syndrome (JPS) was made. As JPS is an autosomal dominant disease
1
and there are no other known cases of JPS in her family, this was felt to likely reflect a de novo mutation. Subsequent colonoscopy demonstrated a mix of both hyperplastic and adenomatous polyps in the cecum and ascending colon. H&E stains of hyperplastic polyp at 10x (A), 100x (B), and 200x (C). Gross surgical specimen at low (A) and high (B) magnification.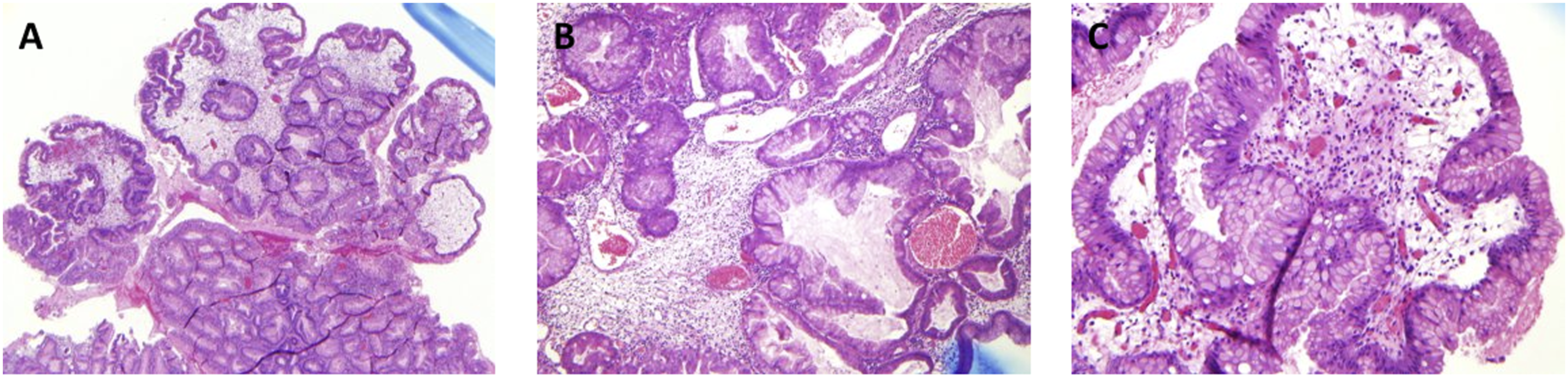
Juvenile polyposis syndrome is caused by monoallelic germline mutations in the SMAD4 or BMPR1A genes and is characterized by predisposition to hamartomatous polyp formation in the gastrointestinal tract, including the stomach, small bowel, colon, and rectum. The term “juvenile” in JPS refers to the pathologic appearance of the polyps rather than patient age of polyp onset; however, most individuals begin to develop polyps prior to adulthood. Diagnostic criteria for JPS include a patient with greater than 5 juvenile polyps in the colorectum, greater than one juvenile polyp elsewhere in the gastrointestinal tract, or any number of juvenile polyps in the setting of a family history of JPS. Identification of a heterozygous pathogenic variant in either SMAD4 or BMPR1A confirms the diagnosis. 1
Up to two-thirds of mutations in JPS are thought to arise de novo, and therefore one should have a low threshold to send patients for genetic counseling and screening when multiple polyps are found in a young patient, even in the absence of family history. While most JPS-associated polyps are benign, malignant transformation can occur. For those with confirmed SMAD4 or BMPR1A mutations, the risk of colorectal cancer development is 20% by age 35 and as high as 65-70% by age 60. 1 For those with gastric polyps, lifetime gastric cancer risk is about 20%. 1
Importantly, patients with heterozygous SMAD4 mutations are also at risk of developing a concurrent syndrome known as Hereditary Hemorrhagic Telangiectasia (HHT) 2 and it is common for such patients to have a combined syndrome of JPS/HHT. HHT is characterized by impaired development of intervening capillary beds throughout the body, resulting in the presence of multiple arterio-venous malformations (AVMs). While HHT can occur due to mutations in the TGFβ signaling pathway including ACVRL1, ENG, or SMAD4, SMAD4 mutation accounts for only 1% of all cases of HHT. The most common clinical presentation of these patients is spontaneous and recurrent epistaxis beginning in childhood. On physical exam, telangiectases can be found on the lips, tongue, buccal and gastrointestinal mucosa, face, and fingers. Larger AVMs can occur in the lungs, liver, or brain. Complications from these larger AVMs including bleeding or shunting, can be sudden, profound, and potentially lethal. In addition to high-output heart failure due to large pulmonary or hepatic AVMs which can result from shunting, Heald and colleagues demonstrated that 23-38% of patients with SMAD4-associated HHT also have aortopathies (including aneurysmal dilation and/or dissection) on transthoracic echocardiogram. 3
In the setting of a patient with both AVMs and intestinal polyps, concurrent HHT/JPS should be considered. While it is unknown how frequently JPS and HHT occur simultaneously, the overall penetrance of HHT due to SMAD4 mutation is predicted to be 95%. It is likely that patients with JPS due to SMAD4 mutation who are not simultaneously diagnosed with HHT may be related to variable expressivity, late-onset of HHT symptoms, or more likely, a clinical evaluation that failed to recognize clinical manifestations of HHT.2,4 A study from O’Malley and colleagues demonstrated that re-examination of SMAD4-mutated JPS patients reveals a large number of patients who manifest symptoms of HHT that were previously unrecognized. In their cohort of 21 patients with SMAD4-associated JPS from 9 families, 81% met diagnostic criteria for HHT and subclinical AVMs were identified nearly universally. 4 While not externally validated, this data suggests that concurrent syndromes of HHT are likely under-diagnosed in SMAD4-mutated JPS patients.
Patients with JPS should be monitored for gastrointestinal bleeding, anemia, abdominal pain, or changes in bowel habits or stool caliber, color, or consistency. Endoscopic surveillance with polypectomy is indicated for patients with confirmed JPS in order to reduce the risk of cancer, bleeding, and intestinal obstruction. 1 In the absence of symptoms, colonoscopy and upper endoscopy should begin at 15 years and repeat annually in the setting of polyps or once every 2-3 years in the absence of polyps. When more polyps than can reasonably be controlled with endoscopic polypectomy are identified, surgical resection with removal of all or part of the stomach/colon may be necessary for either symptom control or to prevent cancer risk. 1
Importantly, for patients with either confirmed JPS/HHT or those who are at risk (SMAD4-related JPS patients), it is important to consider cardiopulmonary complications of HHT including high-output heart failure and physiologically significant aortopathy.2,4 Preoperative evaluation for signs/symptoms of heart failure should be performed for patients with known SMAD4 mutation. Additionally, transthoracic echocardiography may be useful in diagnosing subclinical cardiomyopathy and/or aortopathy which may impact perioperative risk of cardiopulmonary events and decompensations. On skin examination, no telangiectasias were identified on our patient, and formal evaluation for HHT remains ongoing.
Relatives of patients with JPS or JPS/HHT should be screened for JPS-related syndromes. If a genetic mutation is known in the proband, family members should undergo molecular genetic testing. If a genetic mutation is not definitively confirmed in the proband, it is recommended that their family members undergo endoscopy (including upper endoscopy and colonoscopy) as well as blood work (including a CBC), in addition to a thorough history and physical examination, to detect manifestations of JPS. 1 Our patient’s parents and siblings were confirmed to be without genetic mutations or clinical manifestations of JPS, confirming the de novo nature of her SMAD4 mutation. One of her two children were confirmed to carry the pathogenic SMAD4 mutation, and thus will require early surveillance.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.