
Abstract
Several personal descriptions of migraine with aura from 1870 onwards reported a slow, gradual progression of symptoms. Lashley in 1941 meticulously chartered his own auras and concluded that the symptomatology reflected a cortical process progressing with a speed of 3 mm/min across the primary visual cortex. Leão described cortical spreading depression (CSD) in rabbits in 1944 and noticed its similarity to the migraine aura. Despite these scattered pieces of evidence, the prevailing theory was that the migraine aura was caused by a vasospasm and cortical ischaemia. The advent of a technique for measurements of regional cerebral blood flow (rCBF) in 1974 made it possible to detect spreading oligaemia during migraine aura. Between 1981 and 1990 a series of studies of rCBF during migraine attacks showed reduced brain blood flow posteriorly spreading slowly and contiguously anteriorly and crossing borders of supply of major cerebral arteries. These observations refuted the ischaemic hypothesis. The human studies showed initial hyperaemia followed by prolonged hypoperfusion. The relation between aura and CSD was known to cause short-lasting, and therefore not obvious vasodilation and it was considerably strengthened by the demonstration of a long-lasting oligaemia in rats in the wake of CSD. In the primates CSD is not easily elicited, but it has in recent years been clearly demonstrated in patients with brain trauma and stroke. Finally, mutations for familial hemiplegic migraine have been expressed in mice and lower the threshold for CSD. The seminal papers on rCBF and CSD published in the 1980s caused a dramatic shift in our concepts of migraine aura. They moved attention from ischaemia to CSD and thereby to the brain itself, and paved the way for subsequent discoveries of brainstem mechanisms.
Introduction
The migraine aura is clinically dominated by one feature: the gradual spread of symptoms (1–5), usually over 5–20 min for each symptom (6). In contrast, the symptoms of transient ischaemic attacks occur instantaneously and epileptic Jacksonian sensory auras spread over seconds. The slow progression of symptoms suggested to Liveing in 1873 that the symptoms were due to a ‘nerve-storm’ (1). He and many others substantiated their descriptions by actual drawings of their auras (3, 4), but these astute critical observations were forgotten and the age-old vasospastic theory [(7), Marcussen & Wolff 1950] dominated until refuted by regional cerebral blood flow (rCBF) measurement in the 1980s. However, the beginning of the change of concepts had started already in 1941 when the visual physiologist Lashley was able to map the aura retinotopically by drawing his own visual auras. He calculated the speed of the process in the visual cortex to be 3 mm/min (8). In 1944 Leão described the cortical spreading depression (CSD) of the electroencephalogram (EEG) in rabbits (9). Apart from one short note by Milner in 1959 (10) on a possible correspondence between the scotomas of migraine and CSD of Leão, no attention was given to the observations of Lashley and Leão.
From 1967 CBF could be measured during migraine attacks (11). These early studies supported the vasospastic theory with ischaemia as the cause of aura (12) and subsequent hyperperfusion causing the headache (13).
The development of a scintillation camera with 254 channels for use after angiography with intracarotid injections of xenon-133 by the Lassen group (14) was a major technological advance allowing the measurement of rCBF from as many regions of one hemisphere. Serial measurements of rCBF before and during aura triggered by cerebral angiography showed in 1981 a slowly spreading oligaemia over the hemisphere (15) suggestive of a CSD. In migraine without aura (MoA) (so-called common migraine) there were no changes in rCBF (16). The different rCBF patterns were crucial in making the distinction between migraine with and without aura in the International Classification of Headache Disorders from 1988 (6).
In the following, a historical review of rCBF and CSD in the migraine field will be presented, starting with Lashley's detailed description of his auras in 1941 (8). It took 40 years before attention again was paid to the old observation that aura symptoms are slowly progressing (1) and must be due to a special cortical process (2, 8). In a sense, this took us back to the ‘nerve-storms’ of Liveing (1) but at a much higher level of understanding.
This history is an outstanding example of the interplay between clinical human experimental research and basic animal research.
Seminal works of Lashley (1941) and Leão (1944)
Detailed description of migraine aura by Lashley in 1941
In 1941 Lashley (1890–1958), who was a visual physiologist, published some observations of his own attacks of scintillating scotomas (Figure 1). A scotoma usually first occurred as a small blind or scintillating spot in or immediately adjacent to the fovea. Then the spot increased in size and drifted towards the temporal field of one side (8). He had the opportunity to observe and map a large number of scotomas, which were uncomplicated by any other symptoms of migraine. He summarized his observations as follows: ‘Maps of the scotomas of ophthalmic migraine sketched at brief intervals during an attack suggest that a wave of intense excitation is propagated at a rate of about 3 mm. per min. across the visual cortex. This wave is followed by complete inhibition of activity, with recovery progressing at the same rate’ (8).
A visual aura experienced by Lashley (8). The point of fixation is marked by an X. Times in minutes are given below the figures. The blind spot of the opposite eye is marked by a dotted circle. Areas of scotoma are shown alternating with a broken and a full line. Both the lower and upper quadrants are affected, although it is much more in the lower quadrant (5).
Discovery by Leao of cortical spreading depression in 1944 (17)
In his first seminal paper in 1944, Leão (1914–1993) (18) described how EEG activity was successively depressed in different channels depending on their distance from an electric stimulus. An example is shown in Figure 2. The recovery usually took 5–10 min. In addition, evoked activity to touch was depressed (9). In his second paper of 1944, Leão described the reaction of the pial circulation observed with a compound microscope in connection with CSD (19). A wave of marked arterial dilation and increased blood flow in the pial vessels travelled simultaneously with the wave of CSD over the cerebral hemisphere (19). Leão observed occasionally that the period of marked dilation was followed, in some arteries, by a long period of much slighter reduction of calibre (19). In the third paper on CSD from 1945 Leão and Morrison demonstrated that CSD was not inhibited by anoxia (20). They proposed that CSD may be related to migraine with aura (MA) because of the slow development of scotomata and sensory symptoms (20), even if the scotomata were, according to Wolff (7, 21), due to cerebral vasoconstriction. However, Wolff wrote in the 1963 edition of his book on headache (7): ‘This evidence supporting the view that the occipital lope may be implicated in some patients with scotoma as part of the migraine attack and that the defect is due to the cerebral ischemia secondary to vasoconstriction is not conclusive’ (p.239) (7).

Interestingly, Leão did not attempt to calculate the speed of CSD in these first papers (9,19,20). It was later calculated to be 3 mm/min (22). Bures did a lot of work on CSD and demonstrated, for the first time, that CSD can occur in human hippocampus (21). In 1959 Milner stated in a short note that ‘attention should be drawn to the striking similarity between the time courses of scintillating scotomas and Leão's spreading depression because, if there is a true correspondence between these phenomena, there is hope that some of the work done on spreading depression can be brought to bear on the problem of migraine’ (10). No notice was taken of this, probably because the prevailing theory at that time for the aura was that it was caused by cerebral vasoconstriction (7, 21).
Studies of cerebral blood flow in migraine before 1981
O'Brien, using a xenon-133 inhalation technique, reported in 1967 a reduction of about 20% in the cerebral perfusion rate of seven patients during the aura phase of migraine attacks. This was a preliminary publication, and the full paper with 18 patients was published in 1971 (23). One scintillation counter was placed on each side of the headache. Thus only hemispheric CBF was measured. During the aura a 23% reduction in regional cerebral perfusion rates was observed and during the headache phase there was an 8% increase in CBF (23). There was no difference between the two hemispheres, a somewhat peculiar finding (24). The inhalation method has a potential for error in migraine because it cannot distinguish reliably between cerebral and extracranial blood flow (25).
In 1969 Skinhøj (1918–1983) and Paulson used the xenon-133 intracarotid technique with 16 detectors to investigate two patients (26). During the aura phase in one patient there was a reduction in rCBF of up to 66% compared with before the aura, a level known to be critical for normal oxygenation. In the other patient in the headache phase of migraine there was hyperaemia (81 ml per 100 g/min) (26).
Skinhøj used in 1973 the intracarotid xenon-133 clearance method with 16–35 detectors for determination of rCBF within the internal carotid area in four patients during the aura phase of migraine (13). All patients showed severely reduced perfusion, in some areas to a level critical for adequate oxygenation (13). Six patients examined during the headache phase showed significant hyperperfusion. Intracerebral lactate-acidosis was found by spinal tap in all cases. It was suggested that even MoA may be preceded by subclinical cerebral hypoxia (13).
In 1973 a patient with a longstanding severe aura was investigated by Simard and Paulson with the intracarotid technique. A reduced CBF was found and it was unchanged after inhalation of CO2, indicating vasomotor paralysis (27).
Using the intracarotid technique Norris et al. studied a young woman during the aura phase, during the headache phase and after the headache was alleviated by an ergotamine injection (28). The CBF values (ml per 100 g/min) were 38, 63 and 64, respectively, indicating that CBF was not related to headache (28).
Mathew et al. used the intracarotid xenon-133 technique in 1976 and found hypoperfusion during migraine aura in three patients and hyperperfusion in the headache phase in 15 patients (29). On average, rCBF was obtained from 12–15 regions of the hemisphere.
In a prelude to the subsequent Olesen et al. paper (15), Hachinski et al. in 1977 used the intracarotid xenon-133 method with 254 detectors developed by Lassen (1926–1997) et al. (14) in two migraine patients. One patient had an atypical complicated migraine with left leg weakness and left hemianaesthesia and he was investigated on day 5 of this attack. Low CBF values were found and he developed a severe right frontal headache with nausea. He became confused and obtunded, and CBF was now decreased even further with values below the ischaemic level of 20 ml per 100 g/min in large areas of the posterior half of the right hemisphere (25). The important points are the ischaemic rCBF levels and the development of migraine headache during hypoperfusion in this patient. In a patient with MoA there were no changes in rCBF during the development of the attack (25).
Sakai and Meyer using inhalation of xenon-133 concluded in 1978 that cerebral hyperfusion during migraine headaches in 13 patients was mainly due to post-ischaemic reactive hyperaemia but might be augmented by functional hyperaemia because of the head pain itself (13). They failed to reveal any significance differences in the pattern and time course of cerebral hyperperfusion between MA and MoA in both the headache and the post-headache interval (13). This indicated that ‘migraine without aura may also be preceded by a cerebral vasoconstrictive phase even though this is not recognizable clinically as a clear-cut prodrome’ (13).
Seminal works by Olesen et al. (1981) and Lauritzen et al. (1982)
The prerequisite for a more precise characterization of rCBF during migraine was the development by Lassen et al. (14) of a multichannel imaging system with intracarotid injection of xenon-133 and 254 detectors, which resulted in a spatial resolution of 1 cm (30).
Six patients with MA were followed with serial measurements of rCBF with the intracarotid xenon-133 method from the normal state into the aura phase, and in three cases into the headache phase (15). The attacks were initiated by focal hyperaemia in three cases, and during the aura phase all patients developed rCBF reduction (oligaemia), which only in one case approached critical values. Oligaemia gradually spread anteriorly in the course of 15–45 min (Figure 3). In four cases severe headache was present concomitantly with oligaemia. Conclusions from the 1981 paper: ‘The results indicate that the vasospastic model of the migraine attack is too simplistic. Alteration in neuronal function, in the blood-brain barrier, or in some other brain process is more likely to be the primary event of the attack’ (15). CSD of Leão was considered (Lauritzen, personal communication), but CSD was at that time known to be associated with hyperaemia (19, 31). This fact spoke strongly against CSD being the primary neuronal process underlying the spreading oligaemia.
Regional cerebral blood flow (rCBF) in a patient with migraine with aura examined with carotid injection of xenon-133 and 254 detectors in a study by Olesen et al. from 1981(15). An oligaemia starts in the posterior part of the brain and is slowly spreading over the hemisphere.
Inspired by the rCBF results (15), Lauritzen et al. investigated in 1982 CBF in rats during and in the wake of CSD by quantitative autoradiography (32). As shown in Figure 4, cortical blood flow increased 218% during the CSD wave, but, more importantly, it decreased 15–27% after the hyperaemia and for > 1 h after CSD. The changes in blood flow were largely limited to the cerebral cortex. This was the first time that oligaemia had been observed in connection with CSD. The authors concluded: ‘We speculate that the spreading oligemia of migraine with aura may be a phenomenon physiologically related to the finding of an oligemia after CSD’ (29). The finding of initial hyperaemia followed by an oligaemia in connection with CSD was later confirmed in anaesthetized cats (33) and awake and freely moving rats (34).
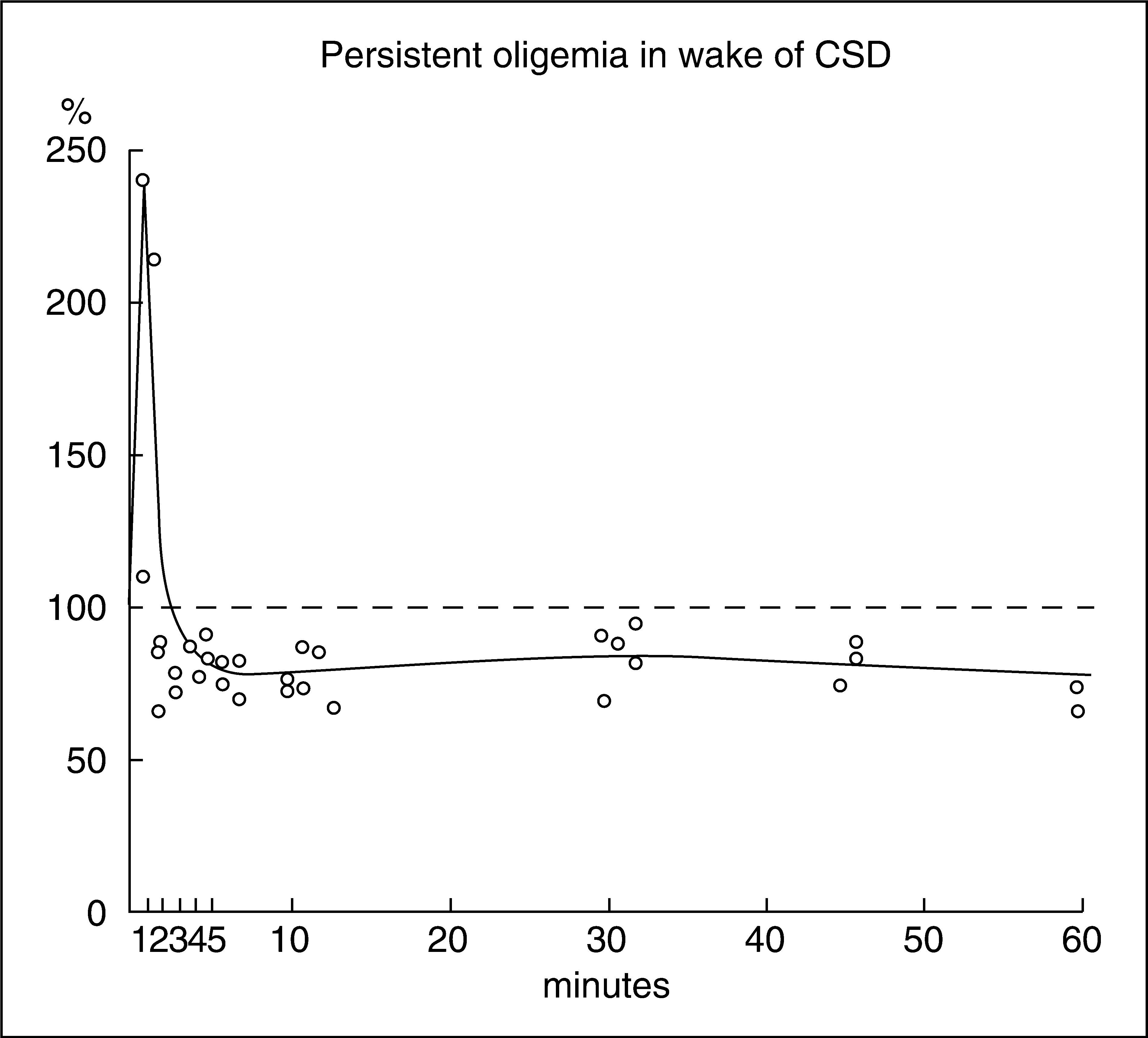
These two papers by Olesen et al. (15) and Lauritzen et al. (32), and their follow-up investigations of rCBF and CSD (24,25,35–39), had an enormous impact on the concept of migraine as a brain disease. For some young investigators these findings were an impetus to join migraine research (Ferrari MD, personal communication).
In MoA (common migraine) there was no change in rCBF (16) and the difference of specific rCBF changes during MA (classical migraine) and MoA was the main reason for dividing migraine into migraine with and without aura in the first edition of the headache classification of the International Headache Society by Olesen et al. from 1988 (6).
Further studies on rCBF and CSD until 1990
The migraine patients in the Olesen et al. paper from 1981 were investigated by coincidence among a series of approximately 250 patients undergoing carotid arteriography (15). The carotid puncture with a catheter placed using Seldinger technique and angiography probably induced the migraine aura (15). In 13 migraine patients who had a carotid angiogram made for diagnostic purposes, rCBF was measured repeatedly at short intervals in order to document the slow spread of hypoperfusion. A wave of reduced rCBF originating in the posterior part of the brain slowly progressed anteriorly with a speed of 2 mm/min (40). Four patients developed headache during the rCBF study at the time of global oligaemia. It was suggested that focal symptoms and rCBF changes might be secondary to CSD of Leão (40).

A new technical development was instrumental for further advance in rCBF measurements in migraine. Lassen et al. developed a safe, atraumatic method, single-photon emission computed tomography (SPECT) using inhalation of xenon-133 for CBF measurements (37). It could be used during spontaneously occurring migraine attacks. Patients from the Copenhagen Acute Headache Clinic, 12 with MoA and 11 with MA, were studied with SPECT (24). In MoA there was no change in rCBF. Of the 11 patients with aura, eight displayed a unilateral region of hypoperfusion (Figure 5), whereas three had normal flow distribution (24). The hypoperfusion remained during the headache phase for up to 6 h. Based on the differences in rCBF changes in MA and MoA, it was again suggested that the two forms of migraine may differ, not only on a clinical basis, but also on pathophysiological grounds.
Repeated single positron emission tomographic examination of regional cerebral blood flow (rCBF) in a patient with migraine with aura. Note the long intervals between measurements. One and a half hours after the start of the aura there is oligaemia in the posterior part of the right hemisphere; after 9 h 30 min there is hyperaemia; and after 21 h there is symmetrical rCBF. (From (24)).
In 1987 Friberg et al. investigated three cases of hemiplegic migraine with the intracarotid method (41). During repeated rCBF measurements all three patients developed spreading focal hypoperfusion originating in the frontal lobe. Instability of cerebrovascular tone (alternating arteriolar constriction and dilation) was observed (41). The authors concluded that instability of cerebrovascular tone may cause focal ischaemia during the course of attacks of classical and hemiplegic migraine (41).
The data of Lauritzen et al. (40) were recalculated together with two additional cases by Skyhøj Olsen and Lassen, who tried to correct the effect of scattered radiation (Compton scatter) on the measured rCBF. They concluded in 1987 that the rCBF reduction that occurred during attacks of classic migraine is sufficient to cause ischaemia and neurological deficits (42) and suggested a vascular origin of the aura symptoms (43, 44). However, this idea was incorrect, partly because of the correction procedure (45) but primarily because it could not explain the typical slow progression of aura symptoms. Later studies with perfusion-weighted magnetic resonance imaging (MRI) have not shown ischaemia during aura (42) and changes in positron emission tomography (PET) and blood oxygen level-dependent (BOLD) MRI have been compatible with a process similar to CSD (46, 47) (see below). The fact that several events during CSD resemble those encountered in cerebral ischaemia led to the question whether CSD in itself induces neuronal injury (48). In 1988 it was found in rats that repeatedly provoked CSDs were not associated with neuronal injury in the otherwise normal brain (48). The energy charge during CSD in rats was normal (49). In man, oxygen consumption was normal during migraine aura despite decreased rCBF (50). These observations are reassuring for the many patients with repeated auras and are compatible with recent studies showing absence of cortical lesions with MRI in patients with MA (51,52).
The longitudinal changes in rCBF were investigated with SPECT and xenon-133 inhalation on admission, after 3–8 h, 20–24 h and 1 week later (37). In seven MA patients, initial rCBF reduction persisted up to 3 h and hyperperfusion was noted later in the previously hypoperfused areas (37). At 24 h rCBF was normal in four patients and hyperaemic in two patients (37). The late hyperaemic asymmetry often persisted beyond the duration of headache. No asymmetry was observed after 1 week.
In 1990 Olesen et al. analysed 10 years of studies of rCBF in migraine (39). Results of rCBF in a total 63 MA patients were reviewed. Twenty patients were investigated with the intracarotid technique (14) and 42 were studied with SPECT (37). The intracarotid method with serial measurements of rCBF provoked MA attacks and the patients usually developed their aura symptoms after one or two rCBF measurements. Focally reduced rCBF was often observed before the patients experienced aura symptoms (39). With SPECT the later phase of spontaneous migraine attacks could be studied. During the headache phase, rCBF gradually changed from abnormally low to abnormally high without apparent changes in headache (39). A summary of these findings is shown in Figure 6 (39).
A summary of the relative timing of regional cerebral blood flow, aura and headaches based on several studies was published in 1990 (39). Note that during aura there is oligaemia and headache occurs during the oligaemic phase and persists into the hyperaemic phase. The hyperaemic phase persists after the migraine headache has stopped, most often after treatment, and is normalized after approximately 12 h. CBF, cerebral blood flow.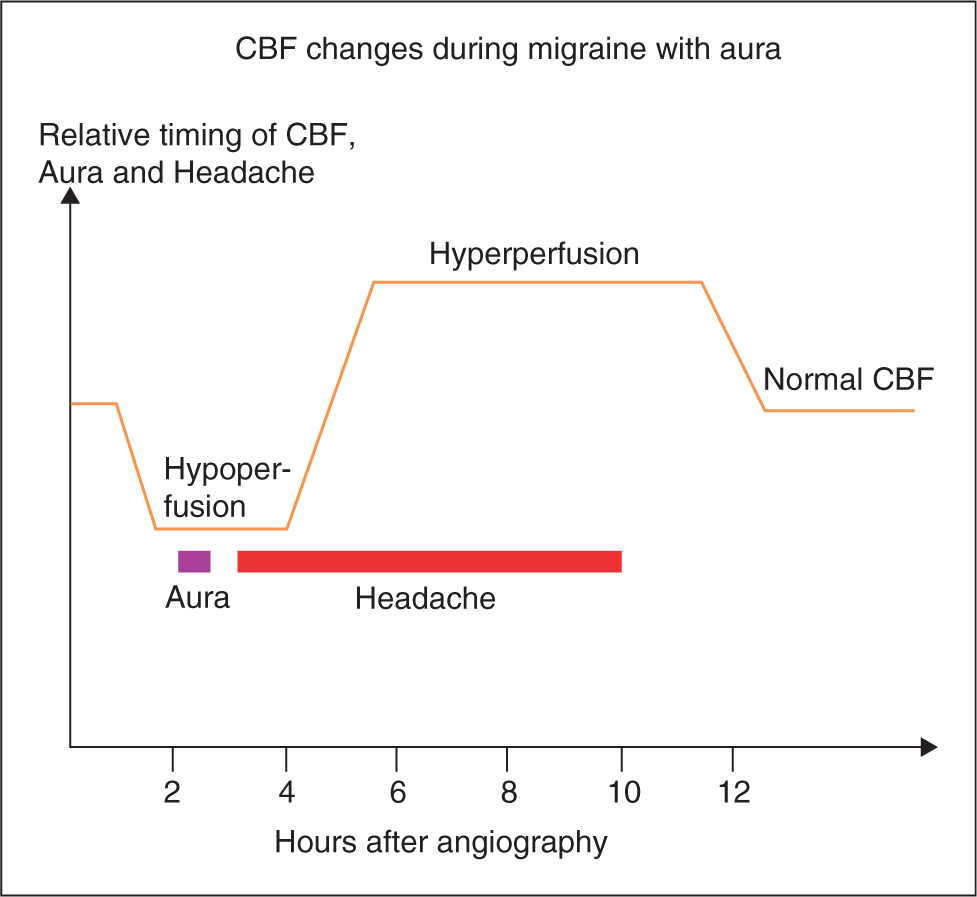
rCBF and CSD after 1990
The literature concerning CSD in animals is overwhelming. For an extensive review see (17). In the following, only animal work with relevance to migraine will be mentioned.
The CSD elicited by KCl significantly increased cell staining (c-fos protein-like immunoreactivity) within ipsilateral trigeminal nucleus caudalis (TNC) (53). This was the first report demonstrating that neurophysiological events within the cerebral cortex can activate brainstem regions involved in pain processing via a trigeminovascular mechanism (53). Late in the wake of CSD in rats a transient hyperaemia occurred (54). This pattern of oligaemia followed by hyperaemia was previously found in MA with SPECT (37).
Weiller et al. investigated nine patients with MoA in a PET study in 1995 by using the C15O2 inhalation technique for the measurement of rCBF (55). No difference in rCBF was found between the hemispheres ipsilateral and contralateral to the headache side, confirming a lack of CSD-like phenomena in MoA. Significantly higher rCBF values were found during the migraine attacks in the brainstem in several planes, slightly contralateral to the headache side (see Figure 7). This increase in rCBF persisted after sumatriptan had induced complete relief from symptoms (55). It has been suggested that this brainstem activation is a kind of ‘migraine generator’ (56), but no evidence supports this contention. In another PET study from 2005 (57), increased lateralized activity in the brainstem was found. Their findings were, however, ipsilateral to the headache side, see below.
Brainstem activation as measured with positron emission tomography in spontaneous human migraine without aura attacks (55). This brainstem activation persisted after subcutaneous sumatriptan had induced complete relief from headache and phono- and photophobia. It has been suggested that this brainstem activation could be a ‘migraine generator’ (59).
Based on turtle cerebellum magnetic field measurements, it was suggested in 1988 that magnetoencephalographic measurements could be used to detect CSD in man (58). Preliminary results obtained with a seven-channel a.c.-coupled magnetoencephalographic system used during migraine were published in 1990 (59), but were not unambiguous (60). In 2001 Bowyer et al. investigated magnetoencephalographic waveforms from patients during spontaneous (n = 4) and visually induced (n = 8) migraine aura (61). Complex d.c. magnetoencephalographic shifts, similar in waveform, were observed in spontaneous and visually induced migraine patients, but not in controls. Two-dimensional inverse imaging showed multiple cortical areas activated in the patients. These results were not inconsistent with a CSD-like neuroelectric event occurring during migraine aura (61). The magnetoencephalographic signals share important features with signals recorded with the same equipment during CSD in rabbits (62). However, the specificity remains to be defined, and further confirmation of the results is needed (60). This future work will probably succeed if selected MA patients can be examined during provoked attacks in an up-to-date magnetoencephalograph.
One patient who had an atypical migraine attack showed bilateral spreading hypoperfusion in a PET study followed by migraine headache (46, 63). The findings were only minimally influenced by scattered radiation (see above) and the study documented beyond reasonable doubt that spreading hypoperfusion is a real phenomenon (63).
Four patients were investigated in 1998 with perfusion- and diffusion-weighted MRI during spontaneous visual aura (42) A 16–53% decrease in relative CBF was observed in the grey matter of the occipital cortex contralateral to the affected visual field. No changes in the apparent diffusion coefficient were observed (42). Eight migraine patients developed atypical visual changes and/or headache during visual stimulation (64). In five patients there was an initial activation pattern as measured with MRI-BOLD (64). This was followed by suppression of activity and the area of suppression progressed across the occipital cortex at a rate of 3–6 mm/min (64). In a later series of cases in 2002 from the same group, migraine attacks triggered by visual stimuli were found in 75% of cases to have increased MRI-BOLD signal in the red nucleus and substantia nigra prior to changes in the occipital cortex, implicating these structures in both MA and MoA (65).
In 2001 Hadjikhani et al., in a landmark study, investigated three patients during visual aura using functional MRI (47). One patient with an exercise-induced aura showed a focal increase in BOLD signal (probably reflecting vasodilation), developed within extrastriate cortex, and this BOLD change progressed slowly (3 mm/min). Then the BOLD signal diminished (possible reflecting vasoconstriction) (Figure 8). This indicated that an electrophysiological event such as CSD generated the aura in the visual cortex (47).
Spreading suppression of cortical activation during exercise-induced visual migraine aura (50). (A) A drawing showing the progression over 20 min of the scintillations and the visual field defect affecting the left hemifield, as described by the patient. (B) A reconstruction of the same patient's brain, based on anatomical magnetic resonance data. The posterior medial aspect of the occipital lobe is shown in an inflated cortex format. A marked perturbation or change in the blood oxygen level-dependent (BOLD) signal was found. It developed sequentially along consecutive regions of calcarine cortex and this perturbation in the BOLD signal was identical from voxel-to-voxel along calcarine cortex, differing only with respect to the time of onset, beginning posteriorly and spreading anteriorly. It was inferred from this study that migraine aura is accompanied by a propagating brain event, which is retinotopically related to the visual perception (moving from central to peripheral visual fields). The changes in BOLD signal were interpreted as, first, an increase in blood flow lasting a few minutes, followed by a longer lasting decrease in blood flow that dropped below baseline levels. The authors concluded that this sequence of events is similar to what is observed during cortical spreading depression and documented repeatedly in rodent or cat cortex (47). MRI, magnetic resonance imaging.
In a rat study from 2001, CSD did not activate neurons in the TNC and did not increase plasma extravasation in the dura mater (66). These findings raised the question whether CSD can cause nociception and headache. In contrast, in a study in rats from 2002, CSD caused a long-lasting blood flow enhancement selectively within the middle meningeal artery (67). This enhancement was dependent upon trigeminal and parasympathetic activation. In addition, there was protein leakage within dura mater. There was increased c-fos expression in the caudal part of TNC. These findings suggested a neural mechanism by which brain events couple to extracerebral cephalic blood flow (67). The results in the two studies are difficult to reconcile, but could be due to different methodology (68, 69).
In a study from 2004, a CACNAIA knock-in mouse was used as a model of familial hemiplegic migraine (FHM). It showed increased susceptibility to CSD (70), indicating that CSD could be involved in FHM.
CSD may alter blood–brain barrier (BBB) permeability by activating brain matrix metalloproteinases (MMPs) (68). Beginning after 3–6 h, MMP-9 levels increased within cortex ipsilateral to CSD, reaching a maximum at 24 h and persisting for at least 48 h. At 3 h after CSD, plasma protein leakage and brain oedema developed contemporaneously. It was concluded that CSD may cause MMP up-regulation and increased vascular permeability in migraine, stroke and trauma (71). However, in 20 migraine patients both with and without aura, no gadolinium enhancement was observed (Sanchez del Rio, personal communication). This strongly indicates an intact BBB during migraine (69).
In 14 patients undergoing neurosurgery after head injury or intracranial haemorrhage, electrocorticographic (ECoG) electrodes were placed near foci of damaged cortical tissue (72). Transient episodes of depressed ECoG activity that spread across the cortex at the rates 0.6 and 5 mm/min were observed in five patients. The rate of progression suggested a CSD (72). In a further similar study from 2006, evidence for CSD or peri-infarct depolarization (PID) was recorded in 50% of patients (71). In a third study, 50% of 63 patients had a CSD after acute cerebral injury (73). Similarly, CSD and/or PID occurred in all but two of 16 patients with large middle cerebral artery infarction (74). These studies from 2002–2008 (71–74) demonstrated beyond doubt that CSD can occur in the human brain.
H2 15O-labelled PET was used to study 24 migraineurs (57). A migraine attack was induced by a glyceryl trinitrate (GTN) infusion. The subjects were scanned pre-infusion, during GTN, during migraine and post migraine. Brainstem activation was seen in the ipsilateral dorsolateral pons. The activation persisted after pain was controlled by sumatriptan. The results suggested that lateralization of pain in migraine may be due to lateralized brainstem dysfunction (57). It is noteworthy that in the first PET study (see Figure 7), significantly higher rCBF values in the brainstem were seen in several planes, slightly contralateral to the headache side (55).
In a study from 2006 the effect of migraine prophylactic drugs on CSD was studied (75). Rats were treated either acutely or chronically over weeks and months with topiramate, valproate, propranolol, amitriptyline or methysergide. Chronic daily administration of migraine prophylactic drugs dose-dependently suppressed CSD frequency by 40–80% and increased the cathodal stimulation threshold, whereas acute treatment was ineffective. Longer treatment durations produced stronger CSD suppression. Chronic D-propranolol treatment did not differ from saline control. The findings suggested that CSD provides a common therapeutic target for widely prescribed migraine prophylactic drugs. Assessing CSD threshold could prove useful for developing new prophylactic drugs and improving upon existing ones.
rCBF changes during and after CSD are not coupled to cerebral metabolism (36) and dilation of cortical surface arterioles propagated with greater intrinsic velocity than the parenchymal CSD wavefront (76). The results suggested a separate mechanism, intrinsic to the vasculature, for propagation of vasodilation associated with CSD (76).
In 2008 Denuelle et al. (77) investigated seven patients with MoA using PET. Measurements were taken during migraine attacks (mean 3 h from onset), after headache relief following sumatriptan injection, and during attack-free interval. They demonstrated a posterior cortical hypoperfusion that was unchanged after sumatriptan. The authors suggested that MoA and MA may have a common pathogenesis (78). It should be noted that in two other PET studies (55, 57) there was no report of posterior cortical hypoperfusion in MoA (see above). This finding, therefore, needs confirmation.
Conclusion
Translational research in migraine has been approached from bedside to bench and from bench to bedside (79). The first step at the bedside was to take a detailed history from the patients (1). This revealed the slow, gradual progression of symptoms specific to the migraine aura (1–4,80). Even if Lashley in 1941 provided excellent proof for a cortical process progressing, with a speed of 3 mm/min, as being the cause of the visual aura (8), and Leão described CSD in rabbits in 1944 (9), it was from 1940 to 1980 the prevailing theory that migraine aura was caused by a vasospasm (7, 21).
The advent of a technique for measurements of rCBF made it possible to detect spreading oligaemia during migraine aura in 1981 (Figure 3) (15). This was literally the end of the vasospastic theory of migraine.
The next step was to go back to the bench in animal studies and show, in 1982, that in the wake of CSD there was a long-lasting oligaemia in rats (32) (Figure 4). The existence of CSD in man has first in recent years been demonstrated after traumatic brain injuries and large cortical infarcts (71–74). Table 1 summarizes the similarity of spreading oligaemia during migraine aura and CSD in rats. Both processes spread slowly with a speed of 2–3 mm/min, there is decreased reactivity to CO2, decreased reaction to functional activation but intact autoregulation. In contrast, autoregulation was impaired in ischaemic, collaterally perfused brain tissue in stroke in man, whereas the CO2 response was preserved (78). The rCBF changes are thus specific for migraine aura and strongly indicative of CSD. Future magnetoencephalographic studies are needed to demonstrate CSD during aura in man.
Finally, it should be noted that rCBF undergoes a sequence of changes during attacks of MA that has not been observed in any other neurological disorder (81).
In the 1950s and 1960s psychological disorders were thought to be a major primary aetiological factor in migraine (82). Even for MA a statement could be: ‘. . . scotomas of migraine are related to previous visual psychological trauma, which are [sic] repressed’ (82).
The two seminal papers from 1981 (15) and 1982 (32) and their follow-up investigations of rCBF and CSD (15,24,25,35,39,49,61,83) had an enormous impact on the concept of migraine as a brain disease.
Footnotes
Acknowledgements
Thanks are due to Professors Jes Olesen and Martin Lauritzen for extensive discussion of the paper. The study was supported by the Lundbeck Foundation via the Lundbeck Foundation Centre for Neurovascular Signaling (LUCENS).