
Abstract
The release of calcitonin gene-related peptide (CGRP) and sensitization of the trigeminal nerve system are important elements in migraine pathophysiology. Sensitization can be induced by topical meningeal administration of inflammatory soup (IS). CGRP release is a marker of trigeminal nerve activation. We examined the effect of intracisternal IS administration on CGRP release in rat jugular vein blood at baseline, 2 and 15 min after the beginning of IS infusion. IS administration caused a significant increase of CGRP levels after 2 and 15 min compared with baseline. Daily oral treatment with topiramate for 4 and 8 weeks led to a dose- and time-dependent reduction of IS-induced CGRP release. Sumatriptan also attenuated stimulated neuropeptide release. These results indicate that intracisternal IS administration leads to activation of the trigeminal system. The inhibition of CGRP release by topiramate offers a possible mechanism that may in part account for the preventative antimigraine activity of this drug.
Introduction
Calcitonin gene-related peptide (CGRP) is a crucial neurotransmitter in the pathophysiology of migraine. For example, newly developed CGRP receptor antagonists (BIBN4096BS/MK-0974) abort migraine attacks in clinical trials (1, 2), as do 5-HT1B/1D receptor agonists (triptans). In experimental animals triptans block stimulus-induced CGRP release in various models, but the evidence for CGRP release and its possible attenuation by sumatriptan during migraine attacks is contradictory (3–5). Nevertheless, blocking of CGRP either at its receptor sites or its release seems to contribute to the abortion of migraine attacks (1, 2, 4, 6).
Among others, antiepileptics and ß-blockers are used for migraine prevention. The mode of action leading to a reduction of migraine attacks is not clear. One current hypothesis is based on the observation that a host of prophylactic substances reduces the probability of cortical spreading depression (CSD) dose- and time-dependently in an experimental rat model (7). In addition, electrical stimulus intensity for the induction of CSD was significantly higher in pretreated animals compared with controls, indicating that preventatives render the brain less susceptible to external stimuli. Acute administration of preventatives was without effect in this study. Besides the suppression of CSD to reduce migraine attacks, other mechanisms may also apply (8–11). For example, preventative antimigraine drugs may act on the release of CGRP (12).
Sensitization during migraine attacks is a phenomenon that has been described extensively in recent years (13, 14). Sensitization can be induced experimentally by topical administration of inflammatory soup (IS) on the dura mater. The effects of IS when administered intrathecally are not known as yet. In addition, the role of CGRP in sensitization is not entirely clear, as topical administration neither sensitized nor activated trigeminal nociceptors (14).
The aim of the study was to investigate the relationship of intrathecally administered IS and the release of CGRP. We hypothesized that intrathecally administered IS could activate the trigeminal nerve system, resulting in CGRP release.
Materials and methods
All experiments were approved by the Landesamt für Gesundheitsschutz und Tierschutz (G0168/05), the body responsible for animal experimentation in Berlin.
Animal procedures
Male Sprague-Dawley rats (240–300 g; Charles-River, Kisslegg, Germany) were anaesthetized with thiopental-sodium (60 mg/kg body weight, intraperitoneally; Trapanal, Altana, Wesel, Germany). When necessary, supplemental doses were administered during the experiments. The rats were placed on a heating blanket and body temperature was maintained at 37 ± 0.5°C using a rectal probe.
The left jugular vein was cannulated (Portex Polythene Tubing, 0.86 mm i.d.; neoLab GmbH, Heidelberg, Germany) for the collection of blood samples and determination of CGRP levels. For the intracisternal administration of IS a soft catheter was introduced into the cisterna magna after a midline skin incision from the occipital protuberance to the cervical area and the respective muscles were carefully moved aside. The flexible catheter (Portex Polythene Tubing, 0.28 mm i.d.; neoLab GmbH) was placed in the cisterna magna and fixed with cyanacrylate adhesive (Contact VA100; Weicon, Münster, Germany). To avoid unspecific effects the animals were left quiet in anaesthesia after surgery for 3 h. A blood sample (1.5 ml) was then obtained from jugular vein for the baseline level of CGRP.
Thirty minutes later, 70 µl IS (histamine 1 mM, serotonin 1 mM, bradykinin 1 mM and prostaglandin E2 0.1 mM; pH 5.5; adapted from Burstein et al.) (15) or vehicle (NaCl 0.9%) was injected slowly (over 5 min) via the intracisternal catheter into the cisterna magna using a Hamilton syringe. Two and 15 min after the beginning of the infusion, blood samples (1.5 ml each) were drawn from the jugular vein. All blood samples were collected without aspiration in prechilled Eppendorf tubes containing ethylenediaminetetraacetic acid disodium salt solution 0.5 M (Sigma Aldrich, Munich, Germany) and the protease inhibitor Aprotinin (Aprotinin from bovine lung, 0.55 trypsin inhibitor units/ml blood; Sigma Aldrich). The samples were immediately centrifuged with 2000 g at 3°C and then stored at −80°C until further analysis. CGRP concentration was determined using a commercially available Rat CGRP Enzyme Immunoassay Kit (SPI-bio, Montigny le Bretonneux, France).
Animals were divided in six treatment groups. Group 1 (n = 6) received an intracisternal administration of IS, whereas group 2 (n = 6) received intracisternal vehicle administration. In sumatriptan-treated animals (group 3), the left femoral vein was cannulated (Portex Polythene Tubing 0.58 mm i.d.; neoLab GmbH) for intravenous (i.v.) infusion. These rats (n = 5) received an i.v. infusion of sumatriptan 300 µg/kg body weight (Imigran®; GlaxoSmithKline, Uxbridge, UK) over 5 min. The infusion was administered 30 min before intracisternal IS administration. Group 4 (n = 4) received daily oral treatment with topiramate 30 mg/kg body weight (Topamax®; Janssen-Cilag, Neuss, Germany) for 4 weeks before surgery. A constant topiramate intake was obtained by pressing topiramate into standard diet for rats (Altromin purified; Altromin, Lage, Germany). No other food was offered to the animals. Groups 5 and 6 received daily oral treatment of topiramate 80 mg/kg body weight (Topamax®; Janssen-Cilag) for 4 (group 5, n = 6) or 8 weeks (group 6, n = 7).
Statistical analysis
Normal distribution of values was calculated with the Kolmogorov–Smirnov test. All values were normally distributed. Differences between the groups were calculated with the unpaired t-test. Differences within one group over time were calculated using the paired t-test. All data are expressed as mean ±
Results
Intracisternal IS administration causes CGRP release
Baseline CGRP concentrations showed no significant differences between the IS group (17.8 ± 3.4 pg/ml) and the vehicle group (16.1 ± 1.5 pg/ml).
Intracisternal IS injection (group 1) caused a significant increase of plasma CGRP concentration in the jugular vein. This increase was significant at the 2-min (120.8 ± 21.4%, P < 0.05) and 15-min (50.6 ± 15.4%, P < 0.05; Fig. 1) time points compared with baseline. In contrast, vehicle administration (group 2) showed no effects after 2 min (2.0 ± 13.1%, P > 0.05) and 15 min (1.9 ± 9.6%, P > 0.05). At both time points IS-treated animals were also statistically significantly different from controls (P < 0.05).

Intracisternal inflammatory soup administration leads to a significant increase of calcitonin gene-related peptide (CGRP) levels within the jugular vein over time at 2 min and 15 min compared with baseline (∗P < 0.05). At the 2-min and 15-min time points CGRP concentrations are significantly increased compared with vehicle (#P < 0.05).
Sumatriptan attenuates IS-induced CGRP release
Intravenous pretreatment with sumatriptan 300 µg/kg body weight (group 3) led to a significantly lower IS-induced CGRP release at the 15-min time point (10.4 ± 5.6%) compared with group 1 (50.6 ± 15.4%, P < 0.05; Fig. 2). At the 2-min time point a clear tendency towards a reduction of CGRP release could be seen, but failed to reach statistical significance [74.1 ± 14.8% (group 3) vs. 120.8 ± 21.4% (group 1) (P > 0.05)].

Sumatriptan 300 µg/kg body weight (i.v.) causes a significant reduction of inflammatory soup-induced calcitonin gene-related peptide (CGRP) increase at the 15-min time point (#P < 0.05). After 2 min a clear tendency to a reduced CGRP release could be seen, but failed to reach statistical significance.
Topiramate administration leads to a dose- and time-dependent reduction of IS-induced CGRP release
Topiramate treatment led to slightly elevated baseline CGRP levels exclusively in group 5 (topiramate 80 mg/kg for 4 weeks vs. IS, P = 0.047). Baseline levels of CGRP were not different between all other treatment groups.
Treatment with topiramate (30 mg/kg body weight; group 4) for 4 weeks showed only a minimal reduction of IS-induced CGRP release compared with IS-treated animals without prior topiramate treatment. No statistical significance was reached.
In group 5 (80 mg/kg body weight for 4 weeks), IS-induced CGRP release was lower at the 2-min (61.8 ± 28.9%, P > 0.05) time point, but reached statistical significance only at the 15-min time point (−0.3 ± 11.4%, P < 0.05; Fig. 3). Oral treatment with topiramate 80 mg/kg body weight over 8 weeks (group 6) attenuated IS-induced CGRP release significantly after 2 min (62.4 ± 14.3%, P < 0.05) and 15 min (4.8 ± 5.3%, P < 0.05; Fig. 3).
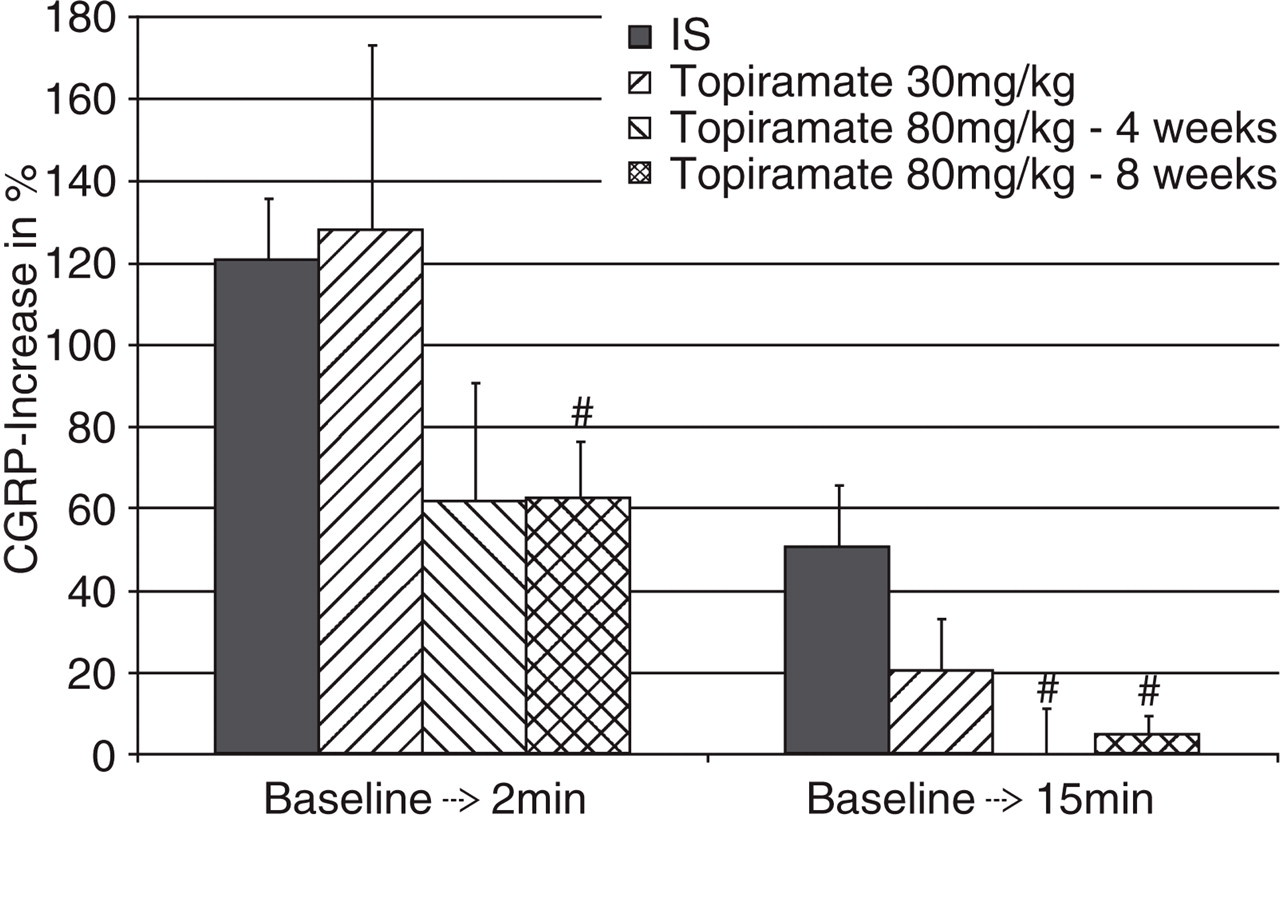
Oral treatment with topiramate results in a time- and dose-dependent attenuation of inflammatory soup (IS)-induced calcitonin gene-related peptide (CGRP) release. Daily treatment with 30 mg/kg b.w. for 4 weeks showed a tendency towards attenuation of CGRP release. Daily treatment with 80 mg/kg b.w. over 4 weeks showed a significant reduction of induced CGRP release at the 15-min time point, whereas the 8 week long daily treatment resulted in clear and significant attenuation of IS-induced CGRP release at both time points (#P < 0.05).
Discussion
We have shown that the intrathecal administration of IS activates the trigeminal nerve system as demonstrated by the release of CGRP. Topiramate blocked neuropeptide release time- and dose-dependently. A single dose of sumatriptan also attenuated CGRP release. The blockade of CGRP release might be a mechanism that contributes to the preventative antimigraine effect of topiramate.
CGRP is a critical neuropeptide in migraine and in experimental models of trigeminally mediated pain. In animal models electrical trigeminal ganglion stimulation as well as superior sagittal sinus stimulation causes the release of CGRP into jugular vein blood, as do many other stimuli (16–18). Blockade of CGRP receptors leads to the inhibition of stimulus-induced vasodilation in dura mater (19).
In some clinical studies CGRP was found elevated during migraine attacks in the jugular vein (3), but this result could not be repeated in a more recent study (5). Many reasons may account for these differences. The most obvious may be the sampling technique with an incomplete number of peptide inhibitors. However, infusion of CGRP leads to a migraine attack in migraineurs (20). Proof of concept for CGRP as a key factor in migraine was achieved when the CGRP receptor antagonist BIBN4096BS successfully aborted migraine attacks in a clinical trial (1). Until now, knowledge about the crucial role of CGRP in migraine attacks results mainly from trials with new CGRP receptor antagonists, but the detailed cascade of action as well as its multiple effects and sites of action remain unclear. Despite all these controversies, CGRP release serves as a marker of trigeminal nerve system activation.
When administered topically onto the dura mater or by i.v. administration in the rat, CGRP does not lead to activation or sensitization of trigeminal nociceptors as demonstrated by single neuron recording in units with Aδ- and C-fibre afferent input in the brainstem (14). These findings raise doubt that CGRP is acting directly on peripheral terminals of nociceptors. Based on experimental data obtained by Storer et al., it seems likely that CGRP serves as a modulating agent of trigeminal activity when administered intrathecally, as the iontophoretic administration of CGRP onto neurons in the trigeminocervical complex led to their excitation (21). Furthermore, the CGRP receptor antagonist BIBN4096BS reduced the activity of trigeminovascular brainstem neurons after systemic CGRP administration (22). So far, CGRP receptor antagonists have not been studied in experimental models of trigeminal sensitization.
Our study was not aimed at demonstrating sensitization. However, we have shown a link between a strong and reliably sensitizing agent and CGRP that has not been identified previously in animal studies of migraine. Zimmermann et al. demonstrated in an in vitro model using a hemicraniectomized skull that proinflammatory mediators (modified IS) cause CGRP release, which is in line with the results of our in vivo study (23). The differences in the amount of CGRP release between this model and our study may be manifold. Among others, model differences between in vivo and in vitro may account for higher CGRP increases in vitro. For example, stimulation thresholds of trigeminal neurons might be lower in the post mortem hemicraniectomized skull and thereby render neurons more susceptible to IS or other stimuli.
The source of CGRP after intrathecal IS administration has not yet been identified. Based on anatomical knowledge of the distribution of CGRP-containing neurons, we assume that one, several or all of the following structures that are components of the trigeminal system significantly contribute to CGRP release into the jugular vein. These are primary dura mater afferents, the afferent trigeminal innervation of the brain vasculature, central processes of trigeminal fibres to the trigeminal nucleus caudalis and nerve terminals innervating the spinal dura mater. Although we have not shown the precise source of CGRP, as it was not within the scope of this work, we hypothesize as follows: we have used a stimulating agent that is well established in models of trigeminal nociception and show the release of a neuropeptide critical in migraine. Moreover, two antimigraine drugs block stimulated CGRP release. In addition IS, CGRP and sumatriptan were clearly linked to the trigeminal system in many experimental studies. Based on this knowledge, we expect that CGRP release due to IS is to a significant extent derived from the trigeminal system.
IS has been used for the study of sensitization in several experimental models of migraine. In these experiments topically administered IS (onto the dura mater) has reliably induced peripheral and central sensitization (13). Moreover, topical administration of IS induces expansion of dural and cutaneous receptive fields (13). Notably, IS also causes enhanced spiking of a central trigeminal neuron when administered on its receptive field within dura mater, indicative of direct stimulation of trigeminal neurons. This effect is significantly less compared with other stimuli such as capsaicin or electrical stimulation of the same receptive field of the dura mater. The latter might be dependent on the concentrations of the ingredients of the IS (24). A comparison of neuronal activity due to other stimuli (e.g. capsaicin) and IS with higher concentrated components has not been published, to the best of our knowledge. The release of CGRP in the in vivo sensitization model has also not been published. In order to compare our results with most recent published animal work of peripheral sensitization, as it relates to migraine, we have chosen the IS components as described there (15).
Our result, that IS activates the trigeminal system, is in line with the above findings indicative of central trigeminal nerve system activation due to IS. It remains to be determined whether IS-induced CGRP release additionally contributes to sensitization of peripheral or central trigeminal neurons. Another open question is the site of action of IS. Two alternatives may apply. IS stimulates either central trigeminal neurons within the trigeminal nucleus caudalis or peripheral meningeal nociceptors due to distribution within the cerebral spinal fluid in the subdural space.
Topiramate is an antiepileptic and preventative antimigraine drug with at least four different targets within the central nervous system. These include blockade of voltage-dependent sodium channels and L-type calcium channels, enhancement of GABAergic neurotransmission, inhibition of glutamatergic transmission and inhibition of carbonic anhydrase. It is not known which of its effects accounts for its antimigraine properties. It has been shown that topiramate—like many other preventative agents—blocks CSD, which is the pathophysiological correlate of the migraine aura (7). Single administration of topiramate was without effect in this study, but repetitive oral administration was highly effective in blocking CSD. A dose- and time-dependent response was shown with the same doses we have used in our experiments. Longer treatment and higher doses led to a significantly reduced probability of CSD occurrence. In contrast, in another study, topiramate also blocked CSD when given once (25). In a study using a trigeminal ganglia cell culture model topiramate blocked potassium-induced CGRP release in a time- and dose-dependent manner without blocking basal CGRP release (12). We have shown that topiramate time- and dose-dependently blocks IS-induced CGRP release. It is not clear whether the reduction of IS-induced CGRP release is due to direct action of topiramate with CGRP synthesis or release or if it is based on the modification of mechanisms upstream of CGRP release such as neuronal depolarization due to IS. One possible mechanism on a cellular basis might be the inhibition of calcium channels by topiramate, as another L-type calcium channel blocker, calciseptine, aborts presynaptic CGRP release in a rat model of neurogenic vasodilation (26).
In summary, our data suggest that topiramate could act not only by elevating the threshold of CSD induction, as demonstrated by Ayata et al. (7), but also by attenuating CGRP release. Both mechanisms separately or combined may lead to the antimigraine activity of this drug.
Acknowledgements
The authors are deeply indebted to Ms Sonja Blumenau for the excellent technical assistance. The project was supported by the Bundesministerium für Bildung und Forschung (BMBF—Project 01EM0515).