
Abstract
The efficacy of a 6-day regimen of frovatriptan for menstrual migraine (MM; attacks starting on day -2 to +3 of menses) prevention in women with difficult-to-treat MM was assessed. Women with a documented inadequate response to triptans for acute MM treatment were included in this placebo-controlled, parallel-group trial. Women were randomized to double-blind treatment for three perimenstrual periods (PMPs) with either frovatriptan 2.5 mg (q.d. or b.i.d.) or placebo initiated 2 days before anticipated MM. The efficacy analysis included 410 women with 85% completing three double-blind PMPs. The mean number of headache-free PMPs was 0.92 with frovatriptan b.i.d., 0.69 with frovatriptan q.d. and 0.42 with placebo [P < 0.001 (b.i.d.) and P < 0.02 (q.d.) vs. placebo]. When migraine occurred, severity was reduced with frovatriptan q.d. (P < 0.001) and b.i.d. (P < 0.001) vs. placebo. Both frovatriptan regimens were well tolerated. In women with difficult-to-treat MM, a 6-day regimen of frovatriptan significantly reduced MM incidence and severity.
Introduction
Approximately half of female migraineurs have migraines associated with menstruation (1), with a 1.7–2.5-fold increase in migraine risk during the period −2 to +3 days relative to the start of menses (2). These attacks may be induced by declining concentrations of endogenous oestrogen before menses or withdrawal of exogenous oestrogen when using hormonal contraception (3–6) and are characterized as being of longer duration, more severe, and more difficult to treat than non-menstrual attacks (1, 2, 7). Triptans (5-HT receptor agonists) are effective for the acute treatment of migraine, including attacks of menstrual migraine (MM) (8–12). Although acute triptan therapy for MM was reported to provide effective pain-free relief at 2 h in most women, a sustained pain-free state was achieved by ≤ 30% of patients (10, 11).
The lack of sustained efficacy in some patients has raised the question of whether better outcomes might be achieved by using triptans to prevent predictable migraines. During acute treatment of migraine, the 5-HT1B/1D agonist activity of triptans inhibits ongoing vasoactive neuropeptide release and the associated vasodilation, neurogenic inflammation and nociception that occur during a migraine attack (13). With respect to MM, achieving and maintaining therapeutic plasma levels of a triptan during the perimenstrual period (PMP), when declining oestrogen levels may precipitate migraine, may help to prevent the attack by binding to 5HT receptors and preventing the vascular and nociceptive components of migraine. Evidence supports a link between oestrogen withdrawal and migraine attacks (4, 14).
In women who do not benefit adequately from acute treatment of MM, short-term (6 days per 28-day cycle) preventive treatment may provide an alternative to acute treatment while avoiding continuous exposure to active drug and the potential for adverse drug reactions associated with daily prophylaxis (14). Studies, including double-blind, placebo-controlled trials (15–18), using short-term preventive (STP) therapy with triptan administration during the PMP have shown promising results. However, no trial to date has specifically evaluated STP in patients with MM attacks that are difficult to treat acutely. The primary objective of the present trial was to evaluate the efficacy of STP frovatriptan in patients who had responded inadequately to one or more triptans for MM attacks over two menstrual cycles and had previously used non-triptan therapy (i.e. were difficult to treat).
Methods
Study design
This Phase IIIb, double-blind, placebo-controlled, parallel-group clinical trial was conducted at 55 sites in Europe and North America from October 2004 to March 2006. The study was designed to evaluate STP treatment of MM with frovatriptan (Frova®; Endo Pharmaceuticals Inc., Chadds Ford, PA, USA) in patients with documented difficult-to-treat MM (see Patients section for definition). MM attacks must have started during the PMP between day −2 to day +3 (inclusive), with the first day of menses counting as day 1 (no day 0) (19). Patients were considered to have MM if they experienced either menstrually-related migraine, defined as migraine with menstruation as well as at other times of the cycle, or pure MM in which migraine occurred only in association with menstruation on or between day −2 to day +3 (19).
The study was divided into two phases. During the single-blind run-in phase, eligible patients were treated with placebo for a single PMP of 6 days. If no MM attack occurred during this PMP, the patient received placebo treatment during a second run-in PMP. Patients were not permitted to use triptan medications to acutely treat MM attacks during the run-in period, unless they were already using frovatriptan for acute treatment. If patients did not experience MM attacks during the two run-in PMPs, they were excluded from entering the double-blind phase. Patients who experienced MM attacks during the single-blind phase were randomized to treat three PMPs with either placebo, frovatriptan q.d. or frovatriptan b.i.d. (see Treatment regimen section). The double-blind phase occurred over a maximum of a 4-month period during which patients took study drug during three PMPs.
The study protocol received Institutional Review Board or Independent Ethics Committee approval and followed the guidelines of the Declaration of Helsinki and the International Conference on Harmonisation for rules on good clinical practice. Written informed consent was given by all patients before enrolling in the trial.
Patients
Women were eligible for this trial if their age was ≥ 15 years (in the USA, France, Sweden and Finland) or ≥ 18 years (in Canada, Norway, Germany, Italy and the UK) and had menses that occurred at regular and predictable intervals. Any women using oestrogen-containing oral contraceptives (OCs) were required to be on a stable regimen that was maintained for a minimum of 2 months before screening. All women had a documented history (≥ 12 months) of MM that also documented MM in at least two of their previous three cycles. Only women with difficult-to-treat MM were included.
Difficult-to-treat MM was defined as having previous exposure to non-triptan (acute and/or prophylactic) therapy for the treatment of MM and an inadequate response to triptan therapy for the acute treatment of MM over a minimum of two menstrual cycles. An inadequate response to triptan therapy was determined by the investigator using the Migraine Medication History Questionnaire and defined as lack of efficacy or poor tolerability, triptan dose in excess of the maximum recommended amount, the need to use rescue medication, recurrence of headache (within 48 h), or partial response. The latter was based upon a ‘Yes’ response to the question ‘Your menstrually-associated migraine headache got better when taking (name triptan), but not as much as you wanted’.
Women were excluded if they were pregnant or breast-feeding; had more than three migraines per month that were not MM attacks or > 15 headache days per month; a history of myocardial infarction, heart disease, coronary vasospasm, peripheral vascular disease, uncontrolled hypertension (> 180 mmHg systolic; > 95 mmHg diastolic), or cerebrovascular disease (including basilar or hemiplegic migraine); severe renal or hepatic dysfunction or any serious illness that would interfere with study participation; or received any investigational medications (within 30 days or 5 half-lives), had a history of allergy to triptans, or had participated in a previous trial of frovatriptan for the prevention of MM.
Randomization and blinding
Patients who successfully finished the single-blind run-in period and continued to fulfil all study criteria were randomized via computer-generated assignments at a 3 : 2 : 3 ratio (q.d. : b.i.d. : placebo). Patients and investigators were blinded to the treatment designation.
Treatment regimen
Patients commenced medication 2 days before the anticipated onset of an MM and continued the therapy for 6 days. On day 1 of treatment, patients received a loading dose of study medication [q.d. group received 5 mg of active drug (2 × 2.5-mg tablets) in the morning and placebo in the evening; the b.i.d. group received 5 mg frovatriptan in both the morning and evening; and the placebo group received two placebo tablets in the morning and evening]. Thereafter, patients received placebo, frovatriptan 2.5 mg q.d., or frovatriptan 2.5 mg b.i.d. on days 2–6. The loading dose and 6-day regimen employed here have been previously shown in pharmacokinetic analyses to provide therapeutic steady-state levels of frovatriptan by day 2 (20). Because MM can begin from day −2 to day +3 (inclusive) of menses, dosing could commence as early as day −4 or as late as day +1. In the latter case, dosing would continue to day +6 from the first day of menses. Additional open-label frovatriptan 2.5-mg tablets were provided (nine per cycle in a separate, non-blinded container) for treatment of breakthrough MM and for non-menstrual (intercurrent) migraine. In the event of breakthrough MM, one additional frovatriptan dose per day could be administered during a PMP, except on the first treatment day when the loading dose was administered. In addition, frovatriptan for breakthrough MM could not be taken within 2 h of study medication. Antiemetics and simple analgesics could be taken for any MM.
Ergotamine-containing preparations and other triptans were prohibited from 24 h before the start of the first dose until 24 h after the end of the last dose of study medication. In addition, a PMP could not be treated unless the person had been free of migraine symptoms for ≥ 24 h and had not taken a triptan (including frovatriptan) or ergotamine-containing preparation within 24 h before the anticipated start of the preventive regimen.
Efficacy assessments
Efficacy data were collected from patient diary cards completed during each PMP. Patients recorded the start date of menses and the time of medication administration. Patients also recorded the occurrence of MM on each day and rated the severity of the attack using a four-point categorical scale recommended by the International Headache Society (IHS) (21). The ratings were 0, no headache; 1, mild/no effect on normal activity; 2, moderate/bothersome but not preventing normal activity; and 3, severe/major disruption to normal activity, bed rest may be required.
Menstrual migraine symptoms (nausea, vomiting, photosensitivity, and phonosensitivity) and functional impairment were rated using a similar scale (0, no symptom/impairment, 1, mild, 2, moderate, 3, severe). Use of any rescue medication for breakthrough MM and the severity of headaches that occurred outside the PMP were also transcribed onto the diary cards. Treatment compliance was evaluated by monitoring the use of medication from returned study blister packs, supplemental frovatriptan bottles, and diary cards. Total migraine burden over all treated PMPs and cycles was calculated as the total number of days with headache pain during a standardized 28-day cycle.
Safety assessments
Adverse events (AEs) were recorded and graded for intensity (mild, moderate or severe). Serious AEs (SAEs) were either life-threatening, required hospitalization for > 24 h, or resulted in significant disability; they were monitored from screening to 30 days after the last dose of study medication. If a patient became pregnant and had a pregnancy complication, that event was recorded as an AE or SAE, as determined by the investigator.
Statistical analysis
Based on previously published data and assuming that response rates for a difficult-to-treat population are proportional to the general population, probabilities were calculated for each group (placebo and q.d. or b.i.d. frovatriptan) that a patient would experience zero, one, two or three PMPs without an MM. The sample sizes for the placebo group and the 2.5-mg q.d. group were then determined using the Whitehead method for ordered categories (22). A power of 90% and a two-sided significance level of 5% were used for the q.d. and placebo groups, and a two-sided significance level of 2.5% was used for the b.i.d. group to adjust for multiple comparisons with placebo. In the placebo and the q.d. dose groups, 150 evaluable patients were required, whereas 100 evaluable patients were required in the b.i.d. group. It was estimated that approximately 600 patients would be screened and 500 patients enrolled to provide 400 evaluable patients.
Two patient populations were defined. The modified intent-to-treat (mITT) population included all patients who received at least one dose of study medication and provided data for the primary efficacy end-point. The safety population included all patients who received at least one dose of frovatriptan (including use as rescue medication) and completed at least one post-treatment safety assessment. The investigational sites were pooled by geographic region or country.
The primary end-point was the number of headache-free PMPs out of three treated PMPs. This end-point was considered a rigorous assessment of the efficacy of frovatriptan, because it required that patients experience no headaches during all 6 days of treatment. The number of headache-free treated PMPs and most secondary efficacy variables were analysed using the Cochran–Mantel–Haenszel test with table scores stratified by pooled investigational site using the mITT population. If a headache was present during the PMP, but some of the data in the PMP were missing, no missing data imputation was calculated. Otherwise, the missing values were replaced by the mean score of the PMPs without missing values. For example, if a patient had one headache in each of two PMPs, with missing data for the third PMP, they then had a mean of one headache per PMP and were scored as having three headaches overall. If they had one headache for one PMP, no headache for one PMP, and were missing data for one PMP, their mean was 0.5 and they were assigned 1.5 headaches overall. A supportive analysis was also performed using a repeated measures logistic model for the primary end-point with pooled investigational site as a covariate and patient as a random effect with an unstructured covariance matrix. An odds ratio (OR) and confidence interval (CI) were generated from this logistic regression. The time to use of rescue medication and time to onset of first migraine were analysed using the multiple-failure time model. Simultaneous comparisons of both doses of frovatriptan with placebo were performed to show efficacy of both doses compared with placebo. Comparisons between frovatriptan doses were not made. To adjust for multiple comparisons, the Holm method was used at P < 0.05 for the first comparison and P < 0.025 for the second comparison. Descriptive statistics were used to summarize continuous variables [n, mean, median, standard deviation (
To assess the occurrence of intercurrent migraine during the postdose interval, two methods were used. In the prespecified analysis, the MM window was based on the patient-determined dosing window and the intercurrent period encompassed all other time points. In this analysis, the active treatments were compared with placebo using repeated measures logistic regression for the incidence of intercurrent migraine across all double-blind PMPs. However, this type of categorization could potentially result in incorrectly classified migraine if a woman dosed prematurely and experienced MM in the postdose period (which was then classified incorrectly as intercurrent migraine). Because of this potential source of error, post hoc analysis using prospectively collected information (i.e. date of menses) was performed using a reclassified 5-day MM window (IHS definition, day −2 to +3) based on actual menses start date. This analysis provides a more accurate account of MM vs. intercurrent migraine, especially because only 34% of women (140/410) accurately predicted onset of menses and dosed within ±1 day of the correct time period in this study. A mixed-effects negative binomial model was used in the post hoc analysis and data from the prespecified and post hoc analyses were compared. All evaluations were performed using
Results
Patient demographics and disposition
Five hundred and eighty-seven women were screened and 427 were randomized to placebo, frovatriptan q.d., or frovatriptan b.i.d.

CONSORT flow diagram of patient disposition throughout the clinical trial. Includes one patient with missing reason for withdrawal. b.i.d., twice daily; mITT, modified intent-to-treat; PMP, perimenstrual period; q.d., once daily.
Demographics among the three treatment groups were similar
Demographics of the safety population

b.i.d., twice daily; MM, menstrual migraine; q.d., once daily;
Reasons for failure of previous acute triptan therapy are listed in Table 2. Over the previous 12 months, an inadequate response was cited for an average of two triptans per patient, and, on average, two reasons for a poor response were given per triptan. The most frequently cited reasons were partial response (range 53–70%), recurrence within 48 h (45–63%) and lack of efficacy (30–47%; Table 2).
Number and percentage of patients previously using triptans acutely by type and by reasons for inadequate response in the difficult-to-treat patient population (N = 416)

Patient is considered difficult to treat if at least one reason for failure of triptan therapy is given; patients can have more than one reason for failure. Patients may have used more than one triptan.
Ninety-eight per cent of patients used at least one concomitant medication during the study, with 93% of patients using antimigraine therapy. Because frovatriptan was provided as rescue medication to all patients, it was the most common antimigraine medication. Non-steroidal anti-inflammatory drugs (74%) and paracetamol (44%) were also used by many patients.
Several concomitant medications with the potential to prevent or ameliorate migraine were also used. Concomitant use of OCs was reported by 35% (placebo), 30% (q.d.) and 24% (b.i.d.) of patients; antidepressant use was reported by 20% (placebo), 16% (q.d.) and 17% (b.i.d.) of patients. Other medications were generally used by < 10% of patients.
During the run-in period, women received placebo during the PMP. Based on data from this period, women subsequently randomized to frovatriptan q.d. may have had somewhat more severe MM during the run-in compared with women subsequently randomized to the other groups
MM characteristics during the placebo run-in period (mITT population)
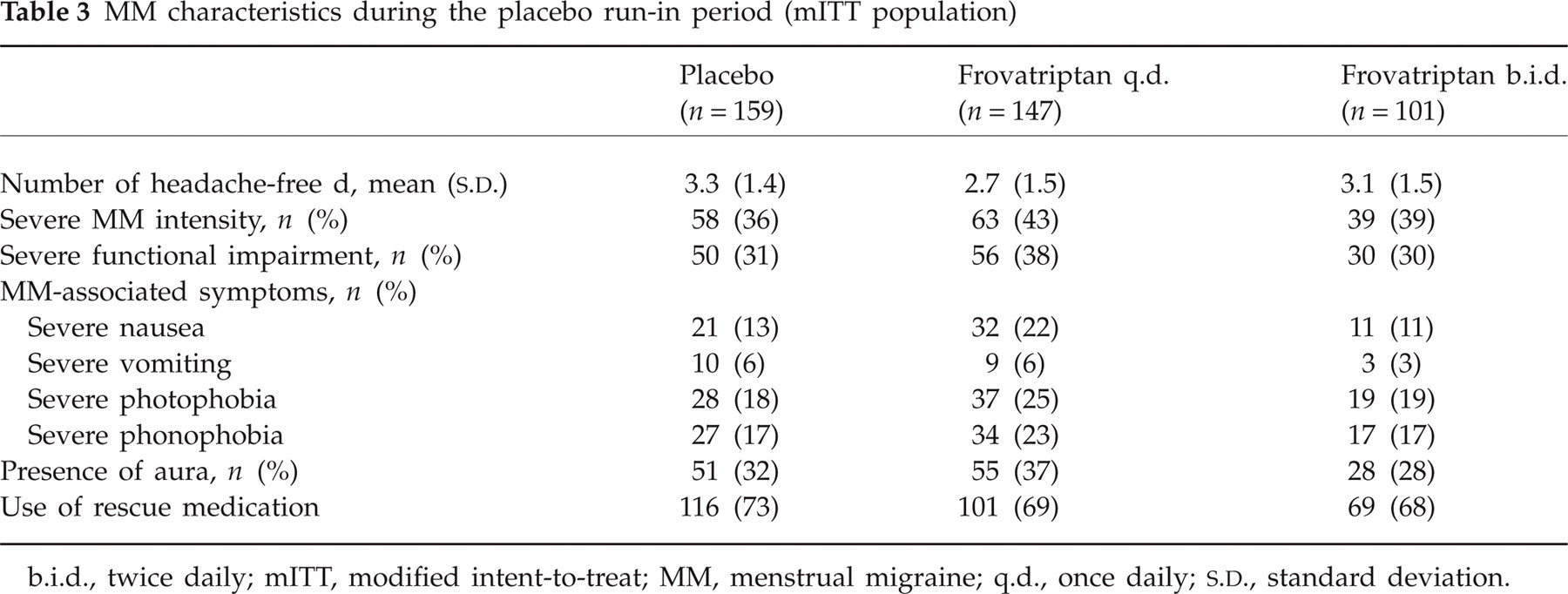
b.i.d., twice daily; mITT, modified intent-to-treat; MM, menstrual migraine; q.d., once daily;
Efficacy
Incidence of menstrual migraine
The mean number of headache-free PMPs per patient (primary end-point) was significantly higher in the two frovatriptan groups [0.69 PMPs (q.d.) and 0.92 PMPs (b.i.d.) compared with placebo (0.42 PMPs) representing 64% (q.d.) and 119% (b.i.d.) increases in the mean number of headache-free PMPs per patient

(A) The percentage of patients who had one, two, or three perimenstrual periods (PMPs) free from menstrual migraine (MM) (modified intent-to-treat population). (B) Percentage of patients who were MM-free during each of three treated PMPs. The total number of patients per dose group (N) and per category (n) are listed. ∗P < 0.05; †P < 0.001 compared with placebo. b.i.d., twice daily; q.d., once daily.
The mean number of MM-free perimenstrual periods per patient

Primary statistical analysis.
Secondary analysis where active treatments were compared with placebo using a repeated measures logistic model with an unstructured covariance matrix.
b.i.d., twice daily; CMH, Cochran–Mantel–Haenszel; MM, menstrual migraine; q.d., once daily; PMP, perimenstrual period;
Use of OCs did not seem to have an effect on the number of MM-free PMPs after frovatriptan preventive therapy; OC non-users had a similar number of migraine-free PMPs to the overall study population with 0.5 (placebo), 0.7 (q.d.) and 1.0 (b.i.d.) MM-free PMPs.
In women who accurately predicted menses onset within 1 day for all three PMPs (n = 140/410, 34%), the mean (
Although 66% of women in the study did not accurately predict MM onset and dose correctly (i.e. per protocol), the overall mITT population still showed a significant decrease in MM days. To explore how mistimed treatment may have affected efficacy, a post hoc analysis was performed in which MM was classified based on menses start; under these conditions women in the placebo group had 52.0% (95% CI 19, 94) more MM days than b.i.d.-treated women over three PMPs (P < 0.001). During PMPs one, two and three women in the placebo group had 33.7% (P = 0.09), 66.9% (P = 0.001) and 57.2% (P = 0.02) more MM days, respectively, than women treated with frovatriptan b.i.d. These results were consistent with the original prespecified analysis, in which the MM window coincided with the patient-decided dosing period, which showed significant differences in MM incidence (PMP 1, P < 0.03; PMPs 2 and 3, P < 0.001 for b.i.d. frovatriptan vs. placebo).
Incidence of menstrual migraine-associated symptoms and functional impairment
The incidence of MM-associated symptoms decreased with frovatriptan compared with placebo (Fig. 3). Before the treatment phase of the study, some degree of functional impairment was reported by 79% (b.i.d.), 85% (q.d.) and 83% (placebo) of patients. For the combined PMPs, the percentage of patients with functional impairment decreased in the frovatriptan groups and was lower compared with placebo, with 78% (q.d.) and 71% (b.i.d.) of patients reporting functional impairment, compared with 93% of placebo-treated patients (P < 0.001; Fig. 4).

The incidence of menstrual migraine (MM)-associated symptoms over all treated perimenstrual periods (PMPs) for the modified intent-to-treat population. ∗P < 0.008 compared with placebo. b.i.d., twice daily; q.d., once daily.

Severity of functional impairment over all treated perimenstrual periods. P < 0.001 for overall functional impairment (combined mild, moderate, and severe) for both frovatriptan groups [78% (q.d.); 71% (b.i.d.)] vs. placebo (93%). Percentages are derived based on the total population per study group; missing values are not represented in the figure. b.i.d., twice daily; q.d., once daily.
Change in the number of headache-free days
During the run-in phase, when all patients received placebo, the mean (
Menstrual migraine characteristics
Frovatriptan decreased the severity of attacks during the three PMPs (P < 0.01; Fig. 5A). The ratio of severe to mild attacks was more than 11:1 in the placebo group, 2:1 in the frovatriptan b.i.d. group and 4:1 in the frovatriptan q.d. group

(A) Maximum intensity of menstrual migraine (MM) pain over all three perimenstrual periods (PMPs). Any missing values were classified as severe. The placebo group had no missing values, and 3% (n = 5) for the q.d. group and 4% (n = 4) for the b.i.d. group were missing. (B) Ratio of severe:mild intensity of MM over all double-blind treated PMPs (modified intent-to-treat population). b.i.d., twice daily; q.d., once daily.
Frovatriptan increased the percentage of patients who experienced no more than a single day of mild headache during the PMP. On average, the number (
Time to first migraine and use of rescue medication
The use of rescue medication in at least one double-blind cycle was reported by 86% (placebo), 67% (q.d.) and 68% (b.i.d.) of women (P < 0.001 for each group vs. placebo). Over all treated PMPs, the hazard ratio for the need to use rescue medication was 0.57 for frovatriptan q.d. and 0.44 for frovatriptan b.i.d. (P < 0.001).
Intercurrent migraine
Intercurrent migraine generally remained the same or decreased in all groups. Over all double-blind cycles, the incidence of intercurrent migraine was 77% in the placebo and b.i.d. groups and 83% in the q.d. group. Kaplan–Meier analysis based on the prespecified analysis of the time to first migraine during the PMP and in the 5 days immediately following dosing revealed some narrowing of the gap between treatments initially, but there was not a sustained trend and the plots remained separated in the 5 days immediately following dosing.

Kaplan–Meier survival curve for the time to migraine onset from the initiation of dosing (days 1–6) during the perimenstrual period (PMP) and for the 5 days following dosing during the (A) second (PMP 2) and (B) third (PMP 3) treated PMPs. b.i.d. = twice daily; q.d. = once daily.
Headache burden
Frovatriptan decreased the total headache burden, or the total number of days with headache pain over a standardized 28-day cycle. Over all three treated PMPs, the total headache burden increased by 0.5 days in the placebo group, whereas the total headache burden decreased by 0.4 and 0.5 days in the two frovatriptan groups (P = 0.05 for b.i.d. compared with placebo).
Safety and tolerability
The incidence of treatment-emergent AEs was similar between groups, with 59% of patients in the b.i.d. and 70% of patients in the q.d. and placebo groups reporting AEs. Twenty-four per cent (b.i.d.), 32% (q.d.) and 19% (placebo) of the AEs were considered possibly or probably related to the drug therapy. However, only 3.8% of patients (n = 16) withdrew because of treatment-emergent AEs (three, eight and four in the placebo, q.d. and b.i.d. groups, respectively); most were during the first PMP of the double-blind period. The most commonly reported AEs were upper respiratory tract infection (37 patients), nausea (36 patients) and dizziness (31 patients).
Patients in the q.d. frovatriptan group reported a higher incidence of migraine (8% vs. 4% for placebo and b.i.d.), headache (7% vs. 3% and 4% for placebo and b.i.d.) and nausea (12% vs. 6% and 8% for placebo and b.i.d.). Most reported AEs were rated mild or moderate in intensity. The incidence of severe AEs was low and appears unrelated to treatment. There were two treatment-emergent SAEs [inguinal hernia, prolonged chest discomfort (8 days)]. Both patients were in the placebo group; however, the patient with chest discomfort had taken frovatriptan as rescue medication 1 day before the SAE. Echocardiogram, ECG stress test and lipid analyses were not significant; there was no evidence of myocardial ischaemia; and the SAE was resolved with ranitidine and, therefore, is likely to have been due to gastrointestinal reflux.
There were no clinically meaningful changes in haematology or biochemistry parameters. Clinically significant changes in 12-lead ECG results were not observed. A more in-depth evaluation of the safety and tolerability of frovatriptan in this population will be discussed elsewhere.
Discussion
In this trial conducted in a population of women with difficult-to-treat MM attacks, the primary end-point and secondary end-points supported the efficacy of frovatriptan for the prevention of MM. The study represents a rigorous assessment of the efficacy of frovatriptan in preventing MM because it was performed in a population of difficult-to-treat women, and it required that women be completely headache free during the 6-day treatment period to meet the primary efficacy end-point. Efficacy was apparent at the first treated PMP, and treatment effects were durable and well tolerated over three double-blind PMPs. These data confirm and extend the observations of the first frovatriptan trial (16) for preventing MM and establish a short-term (6-day) dosing regimen with frovatriptan as a new paradigm in preventing MM attacks.
These results are significant and unique because, although triptan therapies have provided many patients with relief from migraine pain and associated disability, a significant proportion of patients with MM attacks do not derive durable clinical benefits from current therapeutic regimens. In an analysis of 255 women treated acutely with zolmitriptan (n = 119) or almotriptan (n = 136) for MM, approximately one-third of patients did not achieve pain relief 2 h after treatment, < 50% were pain free 2 h postdose, and two-thirds did not report a sustained pain-free response (10). Likewise, in a placebo-controlled trial evaluating the efficacy of sumatriptan for MM, 40% of women did not achieve pain relief within 2 h of taking sumatriptan 100 mg, and only 31% achieved a sustained pain-free response (11). Similar results were reported in a recent clinical trial of zolmitriptan for the acute treatment of MM (23).
Although the above results (10, 11, 23) suggest that a substantial number of women acutely treating MM may fit the definition of difficult to treat, the percentage cannot be estimated because the data only documented an inadequate response during acute treatment of a single MM. In addition, treatment success was defined as a pain-free status from 2 to 24 h. In contrast, the present study enrolled only those women with a history of MM attacks that were classified as difficult to treat over at least two menstrual cycles, and one criterion for this classification was recurrence within 48 h following acute triptan treatment. This population clearly comprised patients with MM attacks that had resisted other forms of treatment but, nevertheless, demonstrated that both doses of frovatriptan significantly increased the number of PMPs that were headache free during the study period.
In addition to preventing headache, the incidence of migraine symptoms (photophobia and phonophobia) was significantly reduced by both b.i.d. and q.d. regimens; nausea was also significantly reduced in the b.i.d. group. Functional impairment was reduced with frovatriptan vs. placebo over all treated PMPs (both P < 0.001). More than one-quarter of patients reported no MM-associated functional impairment after treatment with b.i.d. frovatriptan. Conversely, 54% of patients in the placebo group reported severe functional impairment, and only 37–38% of patients treated with frovatriptan had similar impairment.
As well as reducing the incidence of MM, both doses generally reduced its severity. The net effect of prevention and reduced severity was that frovatriptan-treated patients were approximately two- to threefold more likely to experience no more than a single day of mild MM during the treated PMP. Patients experiencing one or fewer days of mild MM can be expected to retain a near normal functional status, and reductions in headache severity and frequency have been described by patients with frequent headache as being the most clinically meaningful outcomes (24).
The incidence of intercurrent migraine was similar between frovatriptan b.i.d. and placebo, indicating that frovatriptan did not merely postpone the occurrence of a migraine attack. These findings contrast with two recently published trials of naratriptan for STP of MM (18). In both naratriptan trials the percentage of patients who first reported a migraine in the 5 days following the PMP was higher among patients receiving naratriptan compared with placebo, and in both studies the Kaplan–Meier plots of the naratriptan- and placebo-treated patients converged following the treated PMP. Although there is a narrowing of the gap between Kaplan–Meier plots following the PMP, this affect diminishes over multiple treated PMPs and the frovatriptan plots and placebo plot remain separated, which was not reported following treatment with naratriptan. Moreover, the Kaplan–Meier analysis includes the 66% of patients who mistimed their dose relative to menses and MM onset, and therefore experienced postdose MM that was incorrectly scored as intercurrent migraine. When actual menses start dates were used to classify migraines, there was no significant increase of intercurrent migraine (q.d.: RR 1.17, P = 0.13; b.i.d.: RR 1.13, P = 0.31 vs. placebo). At the same time, placebo-treated patients still had 52.0% (95% CI 19, 94) more MM days than b.i.d.-treated women (P < 0.001). Using the IHS definition of MM (using menses start as the reference point), frovatriptan significantly reduces MM without significantly increasing intercurrent migraine.
The analysis of intercurrent migraine over the whole month also addresses the question of whether the long half-life of frovatriptan delayed rebound beyond the length of the Kaplan–Meier analysis. This appears unlikely, because the rate of intercurrent migraine as determined by the prespecified analysis actually decreased in the b.i.d. group; if MM had simply been delayed, there should have been an increase in intercurrent migraine. Moreover, the post hoc analysis in which MM and intercurrent migraine were correctly classified in relation to menses start date confirms this supposition. The absence of an increase in intercurrent migraine and decreases in headache burden also suggest that triptan overuse or abuse was not evident in this trial. It is important to note that patients with more than three intercurrent migraines per month were excluded from the trial; medication overuse would be a potential issue in patients with greater migraine frequency.
Ninety-eight per cent of patients who entered the study were taking concomitant medications, with antimigraine medications being most prevalent (93% of patients). However, even though these patients were taking multiple concomitant medications, the addition of frovatriptan did not change the rate of treatment-limiting AEs. Fewer than 4% of patients withdrew because of AEs. Migraine was listed as the cause for only three patients, all in the q.d. frovatriptan group.
Approximately 24% of women were taking OCs before entering the study. This study was not designed to address specifically differences between those taking or not taking OCs, but the primary efficacy data were numerically similar to the overall population after excluding the OC users from the analysis. Thus, the overall efficacy data did not appear to be affected by inclusion of patients using OCs.
A potential limitation of this study is decreased sensitivity because of (i) patients who could not accurately predict MM onset, (ii) the presence of non-responders, or (iii) use of frovatriptan as rescue medication. It may be that women subjectively assessed their ability to predict an MM, but the characteristics of STP therapy (starting 2 days before the most likely onset of MM and continuing for 6 days), together with the long (26 h) half-life of frovatriptan, should ensure that women generally receive a therapeutic dose during the period of risk. In fact, significant increases in the number of MM-free PMPs were observed among frovatriptan-treated patients in both the overall study population and the 34% of women who predicted the onset of menses within 1 day. The larger treatment effects observed among women who were able to predict the onset of menses (100% improvement for q.d. and 213% for b.i.d.) compared with the overall population (64% for q.d. and 119% for b.i.d.) may be an indication of greater efficacy in this subpopulation. The difficulty some women have in predicting the onset of MM even though they received comprehensive education during the clinical trial suggests that researchers and clinicians need to develop tools that would allow for better prediction of menstruation start and therefore may facilitate better treatment outcomes in menstrual migraineurs.
In addressing the issue of non-responders, it is important to distinguish between an inadequate or non-response to acute triptan treatment vs. preventive triptan treatment. The study was designed to enrol patients who should be classified as non-responders or poor responders to acute triptan treatment, but obviously, some of these patients could show either a non-response or partial response to preventive treatment as well. The observation that when MM occurred it was with significantly decreased severity in the frovatriptan groups indicates a partial response for some patients. Although this partial response is clinically meaningful, a partial response is discounted for the primary study end-point, which requires complete absence of MM over the whole PMP. The net effect of inability to predict the onset of MM and/or being a partial STP frovatriptan responder would be to decrease trial sensitivity and decrease the likelihood of a positive outcome. Despite these limitations, clear positive data were generated with both dosing regimens for the primary and most secondary end-points.
The fact that all patients were provided with rescue frovatriptan would not have had any impact on the primary end-point because a ‘success’ was defined as a completely headache-free PMP. Use of rescue could have affected the outcome of some secondary end-points. However, patients in the frovatriptan groups used significantly less rescue medication, creating a potential bias in favour of placebo. Despite the potential bias favouring placebo, the secondary end-points supported the efficacy of frovatriptan.
Although both dosing regimens of frovatriptan were superior to placebo, the study was not powered to compare doses with each other. When looking at the magnitude of the treatment effects and their statistical significance, the b.i.d. regimen clearly appeared more efficacious than the q.d. regimen. Even with the caveat that the latter group may have been intrinsically more difficult to treat, the superiority of the b.i.d. regimen is consistent with the repeated measures analysis that included severity during the run-in period. The superiority of the b.i.d. regimen is also consistent with outcomes from the previous trial that demonstrated a frovatriptan dose response for STP of MM (16).
Finally, one could question how far the results of this trial can be generalized to the overall population of women with MM. This trial was designed to specifically evaluate women with difficult-to-treat MM, arguably decreasing the chances of showing a beneficial effect, because this population may contain a considerable number of refractory patients. It may be that the large and dose-related treatment effects observed in the first trial assessing STP with frovatriptan are more reflective of the overall population of women with MM because the first trial did not require a difficult-to-treat status for enrolment. Nevertheless, this parallel-arm study provides additional scientific support to the previous crossover trial data by establishing the efficacy of frovatriptan over three PMPs and by focusing on difficult-to-treat patients.
Competing interests
J.L.B. has received clinical research or educational support from Merck, GlaxoSmithKline, UCB Pharma, Allergan, Johnson & Johnson, AstraZeneca, Pfizer, Bristol-Myers Squibb, Winston Laboratories, Sanofi-Aventis, Elan Pharmaceuticals, Novartis, Endo, POZEN, Vernalis, Ortho-McNeil, Advanced Bionics, Forest Laboratories, MedPointe Pharmaceuticals, and Aradigm Corp. M.K. is a speaker and/or consultant for GlaxoSmithKline, Pfizer, Orion, Menarini, Janssen-Cilag, AstraZeneca, MSD Finland, Leiras, and Schering. S.D.S. is an investigator, advisory board member, and/or speaker for Abbott, Advanced Bionics, Advanced Neuromodulation System, AGA, Allergan, AstraZeneca, Endo Pharmaceuticals, GlaxoSmithKline, Medtronic, Merck, Ortho-McNeil, Pfizer, POZEN, ProEthic, and Vernalis. C.P.S. is an investigator for Alexa MDC, Allergan, King, MAP, Merck, Schering-Plough, and Schwartz Biosciences. E.A.M. received research support from Vernalis Development, Ltd. R.S. and J.T. are employees of Vernalis Development Ltd. A.C.P. reports no financial disclosures.
Acknowledgements
The authors thank Dr Richard B. Lipton (Albert Einstein College of Medicine, Bronx, NY, USA) for his comments and careful review of the manuscript. We also appreciate the editorial support provided by Kevin Ryder, PhD, Kristine W. Schuler, MS, and Dana Franznick, PharmD of Complete Healthcare Communications, Inc. (Chadds Ford, PA, USA). This research was supported by Vernalis Development Ltd, Winnersh, UK, and Endo Pharmaceuticals Inc., Chadds Ford, PA, USA.
The Frovatriptan Study Group
Canada: M. J. Gawel, Sunnybrook and Women's College Health Sciences Centre, Toronto, ON; L. D. Sitwell, Riverside Headache Research, Ottawa, ON; R. A. Purdy, Queen Elizabeth II Health Sciences Centre, Halifax, NS.
Finland: M. Kallela, Helsinki Headache Centre, Helsinki; E. Säkö, Turku Headache Clinic, Turku; M. L. Sumelahti, Neurologist Tammer-Tutka, Tampere; T. Jolma, Pori laakarikeskus, Pori Turku Headache Clinic/Pori laakarikeskus, Pori.
France: G. E. A. Geraud, CHU Rangueli, Groupe Hospitalier Rangueli-Larrey CHU Service de Neurologie, Toulouse; M. Lanteri-Minet, Departement d'evaluation et Traitement de la douleur CHU Hopital Pasteur, Nice Cedex; C. Sereni, Hôpital Léopold Bellan, Paris.
Germany: H. Jaeger, Neue Grosse Bergstr, Hamburg; V. Pfaffenrath, Leopoldstr, Munchen; R. Schellenberg, Institut für Ganzheitliche Medizin und Wissenschaft, Huttenburg.
Italy: G. Comi, Centro Cefalee, Clinica Neurologica DIMER, Milano; F. Facchinetti, Dipartimento Integrato Materno Infantile Struttura Complessa di Ginecologia e Ostetricia, Modena; G. Nappi, Centro interuniversitario cefalee e disordini adattativi, Istituto Neurologico C., Pavia.
Norway: A. N. Dueland, Sandvika Neurologpraksis, Sandvika; A. C. Poole, Sjolyst Medical Center, Oslo.
Sweden: C. H. Dahlof, Sodra Vagen, Gothenburg; G. Samsioe, Kvinnokliniken, Lund; A. Ehrenborg, Sodra Torggatan, Kungsbacka.
United Kingdom: E. A. MacGregor, The City of London Migraine Clinic, London.
United States: J. L. Brandes, Nashville Neuroscience Group, Nashville, TN; M. L. Diamond, Diamond Headache Clinic, Chicago, IL; A. H. Elkind, Elkind Headache Center, Mount Vernon, NY; J. Goldstein, San Francisco Clinical Research Center, San Francisco, CA: W. L. Harper, Wake Research Associates, Raleigh, NC; R. P. Hull, North Alabama Neuroscience Research Associates, Huntsville, AL; J. A. Klapper, Colorado Neurology and Headache Center, Denver, CO; N. T. Mathew, Houston Headache Clinic, Houston, TX; A. Mauskop, New York Headache Center, New York, NY; J. S. Meyer, Cerebrovascular Research Laboratories, Houston, TX; L. Mueller; University Headache Center, Moorestown, NJ; A. A. Pragalos, Community Research, Cincinnati, OH; C. P. Schreiber, Headache Care Center, Springfield, MO; S. D. Silberstein, Thomas Jefferson University, Philadelphia, PA; R. P. Singer, Neurology Clinical Research, Plantation, FL; T. R. Smith, Mercy Health Research, Chesterfield, MO; S. J. Steen, Axiom Clinical Research of Florida, Tampa, FL; S. J. Tepper, The New England Center for Headache, Stamford, CT; A. B. Vasquez, Suncoast Neuroscience Associates, St Petersburg, FL; R. L. Von Seggern, PharmQuest, Greensboro, NC; D. S. Roby, Temple University, Philadelphia, PA; M. R. Stein, Neurological Research Institute of the East Bay, Walnut Creek, CA; H. L. McDaris, Medical Affiliated Research Center, Huntsville, AL; S. G. Raj, Benchmark Research, Metairie, LA; T. D. Rozen, Michigan Head-Pain and Neurological Institute, Ann Arbor, MI.