
Abstract
No new preventive drugs specific to migraine have appeared for the last 20 years and existing acute therapies need improvement. Unfortunately, no animal models can predict the efficacy of new therapies for migraine. Because migraine attacks are fully reversible and can be aborted by therapy, the headache- or migraine-provoking property of naturally occurring signalling molecules can be tested in a human model. This model has predicted efficacy of nitric oxide synthase inhibition and calcitonin gene-related peptide receptor blockade. The pharmaceutical industry should pay more attention to human models, although methods are different from normal target validation.
Introduction
Migraine is the most prevalent neurological disorder with an estimated 43 million sufferers in Europe (1) and slightly smaller numbers in the USA (2). It is a very painful condition that often leads to absenteeism from work and more often to considerably decreased efficiency at work. The estimated cost of migraine in Europe is €27 000 million per year which, among the neurological disorders, ranks third after dementia and stroke (1). Migraine is considered to be a disorder of neurovascular transmission without structural lesions. In theory it is therefore fully treatable if sufficiently effective and specific drugs could be found (3).
The advent of the triptans, 5-hydroxy tryptamine (5-HT)1B/D receptor agonists, > 15 years ago (4, 5) helped to throw light on this neglected and often stigmatizing disorder (6). The relatively high efficacy (7) of drugs (triptans) with such specific mechanisms of action suggested that specific neurobiological mechanisms must cause the migraine attack. Furthermore, this class of drugs greatly increased the interest of the pharmaceutical industry in migraine, and boosted a market that had previously been dominated by generic drugs such as ergotamine (8) and dihydroergotamine or by drugs developed for other indications like β-blockers (9, 10). Total global sales of triptans are presently around $3 billion dollars a year. Despite these large sales, migraine has a huge unmet need for a better orally available drug. Thus, only 10% of migraine patients in a Danish county were treated with triptans (11). It is therefore time to consider how existing drugs have been developed and how new targets can be identified that can lead to more effective treatments with fewer side-effects.
The development of migraine drugs
The first drugs for migraine were simple analgesics with no migraine-specific mechanism of action such as aspirin, which is still widely used (12). Next, ergotamine was isolated from ergots in 1918 (13) and introduced in migraine therapy in 1925 because of its presumed sympatholytic activity (14). At that time a school of thought had proposed that migraine was due to sympathetic overactivity, a hypothesis later discarded. The effect of ergotamine was next ascribed to its vasoconstrictor activity (15) and finally to its 5-HT1B/1D and 5-HT2 agonistic activity (16, 17).
The modern era of migraine treatment started with the synthesis of the potent 5-HT antagonist methysergide from LSD25 by Sandoz in Basel (18). Sicuteri from Italy found it very effective in migraine prevention (19). Sicuteri then investigated the metabolism of serotonin and showed that the excretion in the urine of 5-hydroxyindoleacetic acid, the metabolite of 5-HT, was increased during migraine attacks (20). Subsequently, 5-HT was found to be decreased in blood during a migraine attack (21–25) and infusion of 5-HT was shown to relieve migraine attacks (26–28). The human studies focusing on the importance of 5-HT inspired the development of the first triptan, sumatriptan, by P. Humphrey of Glaxo (29). In this work he was also inspired by the finding of Saxena of an ‘atypical’ receptor being responsible for the vasoconstrictor effects of methysergide in the carotid vascular bed studied with the arteriovenous shunt model (29–31).
Animal models of migraine
In the following we shall consider four animal experimental models that have significantly contributed to our understanding of the neurobiology of migraine and other headaches.
In the neurogenic inflammation model of Moskowitz, electrical stimulation of the trigeminal ganglion causes dural protein extravasation (32–34). This model has greatly increased the understanding of the role of peripheral trigeminal nerve fibres around blood vessels. Thus, it has been suggested that during migraine an unidentified stimulus prompts the trigeminal nerve fibres surrounding the blood vessels to release stored peptides and subsequently cause aseptic inflammation of the blood vessels (35). Triptans bind to 5HT1D and 5HT1F receptors presynaptically, hyperpolarize the terminals and thus block neurotransmitter release (36). Dihydroergotamine (36) and non-steroidal inflammatory agents (37) also prevent the stimulation-induced leakage of plasma proteins within the dura mater. The neurogenic inflammation model has been used as an assay to predict probable antimigraine efficacy without causing vasoconstriction. Several such substances have been found: NK1 antagonists RPR100893, LY303870, GR205171 and MK-0869 (38, 39), 5HT1D selective agonists PNU 142633 (40), CP-122,288 (a highly potent inhibitor of neurogenic plasma extravasation in animal models) (41) and endothelin 1B receptor antagonist bosentan (42). All of these compounds could block neurogenic inflammation but failed to relieve migraine in clinical trials (for review, see (43)).
Another model, developed by P. Goadsby, may perhaps be more predictive, although this remains to be demonstrated prospectively. It involves stimulation of cerebral blood vessels in the cat, particularly the superior sagittal sinus. Neuronal activity in the brainstem is recorded. This model did not respond to some of the above-mentioned compounds that failed in clinical trials of acute migraine (44), but it responded to lipophilic triptans naratriptan (45), eletriptan (46) and and zolmitriptan (47), but not sumatriptan (47, 48), to the calcitonin gene-related peptide (CGRP) antagonist BIBN4096S (49) and to ergotamine and dihydroergotamine (50). The model is resource demanding with low throughput and not suitable for screening many compounds. No drugs have yet been taken into man on the basis of results obtained with this model.
The third model was introduced by Strassman and colleagues (51). Their research took Moskowitz's work a step further, showing that when inflammatory molecules were administered to the dura of rats, neurons in the trigeminal ganglion were both activated and sensitized. Furthermore, the model suggested that sensitization of peripheral and central trigeminovascular neurons plays an important role in the pathophysiology of migraine pain (52). Moreover, it has been shown that triptans may exert their antimigraine action through presynaptic 5HT(1B/1D) receptors in the dorsal horn by blocking synaptic transmission between axon terminals of the peripheral trigeminovascular neurons and cell bodies of their central counterparts (53). The major impact of these studies on clinical practice was that triptans seem to have no effect on the hypersensitized state of central neurons clinically presented by cutaneous allodynia in 70% of patients (54). Although promising, the model has not yet been used to validate new treatments for migraine and has not yet generated specific targets for the development of such drugs.
The closed cranial window model in rats preserves the natural environment around pial and dural arteries because a thin, translucent layer of bone is left in situ (55, 56). The diameter of dural and pial arteries as well as laser Doppler recordings of cortical flow can be recorded continuously (57). This intra-vital model has contributed new knowledge about the pharmacology of cerebral and extracerebral arteries. Agonists as well as antagonists and new chemical entities can be evaluated for their vascular effect (56, 58–62). However, the relevance to migraine of the observed effects remains to be demonstrated.
Human models of migraine
Currently, three principally different targets are being explored in the treatment of acute migraine attacks. All have been validated in proof of concept studies and all have been developed primarily on the basis of human studies.
Cortical spreading depression (CSD) was first demonstrated as an animal experimental phenomenon in 1944 by Leao, who also suggested that it was relevant to migraine (63). However, his proposal generated little interest and was virtually forgotten. In the 1980s a series of studies of human regional cerebral blood flow (rCBF) during migraine with aura showed pathognomonic change in rCBF typical of CSD (64–67). Starting at the posterior pole of the brain, an area of decreased rCBF gradually spread forward at a rate of 2–3 mm/min. This progression and some other rCBF findings led to revived interest in CSD as a cause of migraine aura. Newer techniques have recently confirmed human rCBF changes during human migraine attacks that are very similar to CSD (68, 69). These results stimulated A. Parsons, then at Smith Kline Beecham, and his team to developed tonabersat, a compound with no known binding site, but with the ability to block CSD efficiently in animal experiments (70, 71). This compound was first tried for acute migraine attacks, although its profile was much more suitable for prophylactic use. It failed in the acute studies, but has recently shown a tendency (P = 0.14) to a better response than placebo in a migraine prevention trial (72).
Nitric oxide (NO) involvement in migraine has been analysed in a series of provocation experiments using nitroglycerin, glyceryl trinitrate (GTN), as an NO donor (73–75). GTN is highly lipid soluble and easily penetrates membranes including the blood–brain barrier. It can therefore deliver NO to extra- as well as intracerebral compartments. It was first shown that GTN induces headache in normal volunteers in a dose-dependent fashion with a ceiling effect at a relatively modest dose (76). In a series of studies (76–79) this model was refined and an infusion model using 0.5 μg kg−1 min−1 for 20 min was validated. In migraine patients compared with normal subjects infusion of GTN causes a more intense immediate headache during the infusion and, more importantly, it also leads to a delayed headache that is maximal around 6–7 h after the infusion (75). This delayed headache has the characteristics of typical attacks of migraine without aura. Likewise, patients themselves find that the provoked attacks are identical to their spontaneous attacks. In normal subjects delayed headache is largely absent. A non-selective nitric oxide synthase (NOS) inhibitor, N(G)-mono-methyl-L-arginine (L-NMMA), was effective in treating spontaneous migraine attacks (80). This confirmed that NO is an important offending molecule throughout the duration of a migraine attack. L-NMMA is not suitable as a drug because it has poor bioavailability and a short duration of action, and because blocking endothelial NOS increases systemic blood pressure. The above studies greatly stimulated the pharmaceutical industry to develop selective NOS inhibitors. Most recently, a selective inducible NOS (iNOS) inhibitor, GW274150, has entered clinical Phase II trials ( http://www.clinicaltrials.gov/). GW274150 is a highly selective inhibitor of iNOS and offers the potential of anti-inflammatory activity in migraine through a novel mechanism of action (81). The efficacy of neuronal NOS (nNOS) inhibition is not yet known and just recently a selective nNOS inhibitor entered Phase I trial (J. Andrews, communication at International Headache Research Seminar, Copenhagen, March, 2007). Histamine also causes more headache in migraine sufferers than in non-sufferers (82) and a delayed headache that is identical to the spontaneous migraine attacks of the patients (82). The mechanism of action of histamine was via the H1-receptor because mepyramine was able to prevent histamine-induced headache and migraine completely, whereas placebo was ineffective (83). However, it was already known at the time that histamine H1 and histamine H2 blockers are ineffective in migraine prevention. This example showed that, because a substance can induce migraine in migraine sufferers, it does not necessarily follow that blocking its receptors is effective in the treatment of spontaneous migraine. Intuitively, it is understandable that some provoking factors are involved in spontaneous attacks and others not.
CGRP antagonism is the third proven target. CGRP was detected in perivascular nerves of the brain and the dura mater and its receptors were located to vascular smooth muscle in those tissues (84–86). It was shown that CGRP is a very strong vasodilator and, interestingly, more in the cranial than in the abdominal arteries, where it does not induce relaxation (87). On the basis of such studies it was suggested that CGRP may be involved in migraine (Box 1, Fig. 1). Human studies then demonstrated an increase in CGRP in external jugular venous blood during migraine attacks (88). That finding trigged interest in CGRP antagonism as a potential target for antimigraine drugs, but a later study (89) could not confirm this finding. Menawhile, the interest in CGRP antagonism was augmented because infusion of CGRP intravenously to migraine sufferers triggered a migraine-like headache in eight out of 10 individuals and an attack fulfilling migraine criteria in three out of these eight individuals (90). It was thus clear that, similar to NO and histamine, CGRP was present in structures relevant to migraine and that the substance could induce headache and migraine in sufferers. However, would antagonism be effective in spontaneous attacks like NOS inhibition or could antagonism be totally ineffective like histamine receptor blockers? This issue was clearly answered by a Phase II clinical trial of the Boehringer Ingelheim compound BIBN4096BS (91), now called olcegepam. It showed a dose-dependent response in acute migraine with a possible ceiling effect around a dose of 5 mg (92). This proof of concept trial was done with intravenous slow injection. The compound was not taken forward because it is not absorbed by the oral route. Fortunately, other pharmaceutical companies continue to work on oral CGRP receptor antagonists, and at least one such compound was effective in a Phase II trial (93) and is now in Phase III.
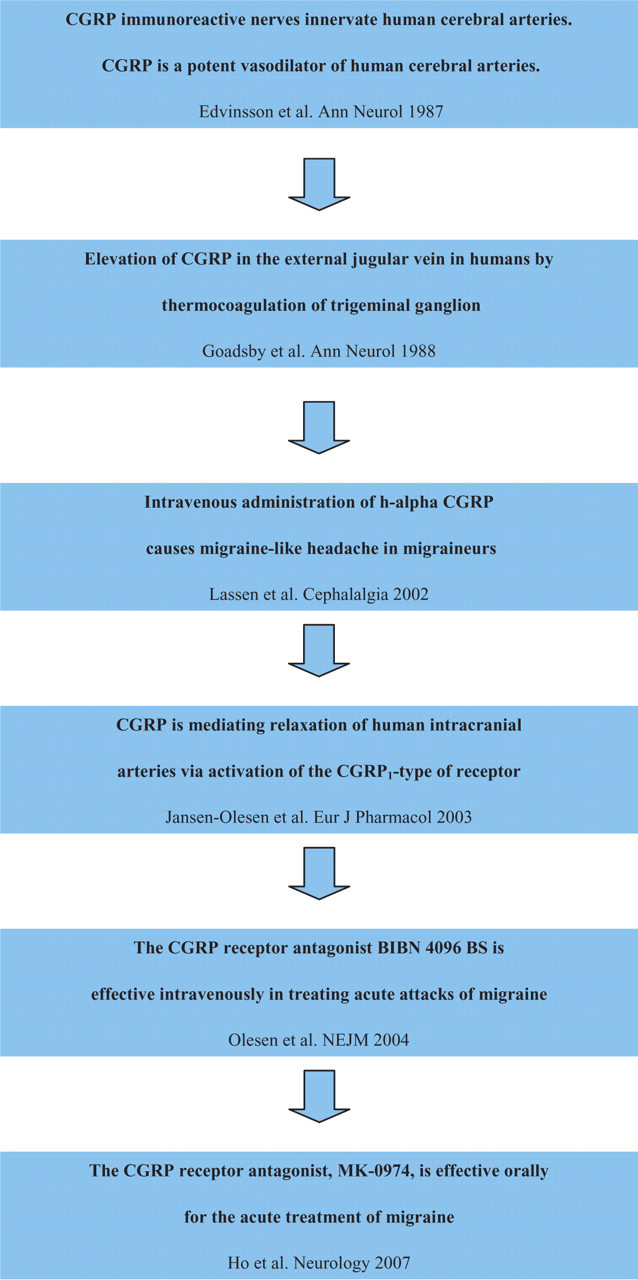
Chronology – the discovery of calcitonin gene-related peptide (CGRP) receptor antagonist for the acute treatment of migraine (key references are shown).
Box 1
CGRP is a 37-amino-acid neuropeptide, identified in 1982 (106). It belongs to a family of peptides including adrenomedullin, amylin and calcitonin with diverse biological functions in the periphery and in the central nervous system (107, 108). Rosenfeld and colleagues (109) reported the presence of CGRP immunoreactivity in the rat trigeminal ganglia. Mason et al. (110) demonstrated calcium-dependent CGRP release upon depolarization from trigeminal ganglia in vitro. These studies provided the first evidence of possible CGRP involvement in migraine. A dense supply of CGRP-containing fibres around the cerebral vessels was reported to originate in the trigeminal ganglion (111). This was the first report of CGRP involvement in the trigeminovascular reflex. CGRP-containing nerve fibres innervate human cerebral arteries. Furthermore, CGRP is a potent vasodilator of human arteries (112) and mediates relaxation of these arteries via activation of the CGRP (1)-type receptor (113). In 1988 Goadsby et al. (114) reported that CGRP was released into the extracerebral circulation of humans during thermocoagulation of the trigeminal ganglion. Studies in migraine patients showed elevation of CGRP during (88) and outside of migraine attacks (115). However, a recent study challenged these reports, showing no changes in plasma CGRP during migraine attacks compared with outside of attacks (89). Despite this controversy, the importance of CGRP in migraine became firmly established. First, Lassen and colleagues demonstrated that intravenous infusion of CGRP may cause migraine-like headache in patients with migraine (90). Second, the CGRP receptor antagonist olcegepam (BIBN4096BS) was effective given intravenously in the treatment of acute migraine attack (92). Recently, an oral formulation of a CGRP receptor antagonist was also effective in the treatment of acute migraine (93).
Target identification in migraine
Animal experiments are of course always necessary to demonstrate the presence of signalling molecules, receptors, ion channels or enzymes in tissues relevant to migraine. So far, the interest concentrates on cranial blood vessels, their perivascular nerve terminals, the trigeminal ganglion and the trigeminal nuclear complex in the brainstem, particularly the nucleus caudalis. It is also useful to know whether a substance dilates cranial vessels. So far all migraine-provoking agents have had vasodilating properties.
To validate a target two different avenues can be pursued. Optimally, the validation of the target should be done in human experiments. This is possible with all naturally occurring substances and with chemical entities that have passed the necessary toxicological studies.
For this purpose we have validated and extensively used a human provocation model in normal volunteers (94, 95) and in migraine sufferers (90, 96, 97). Our present experience is that normal volunteers should be used in the first place (95, 98–100). Only substances that produce substantial headache in healthy volunteers have the potential to induce migraine in migraine sufferers and only such substances should therefore be tested in migraine sufferers. The model is described in Fig. 2 (Box 2).
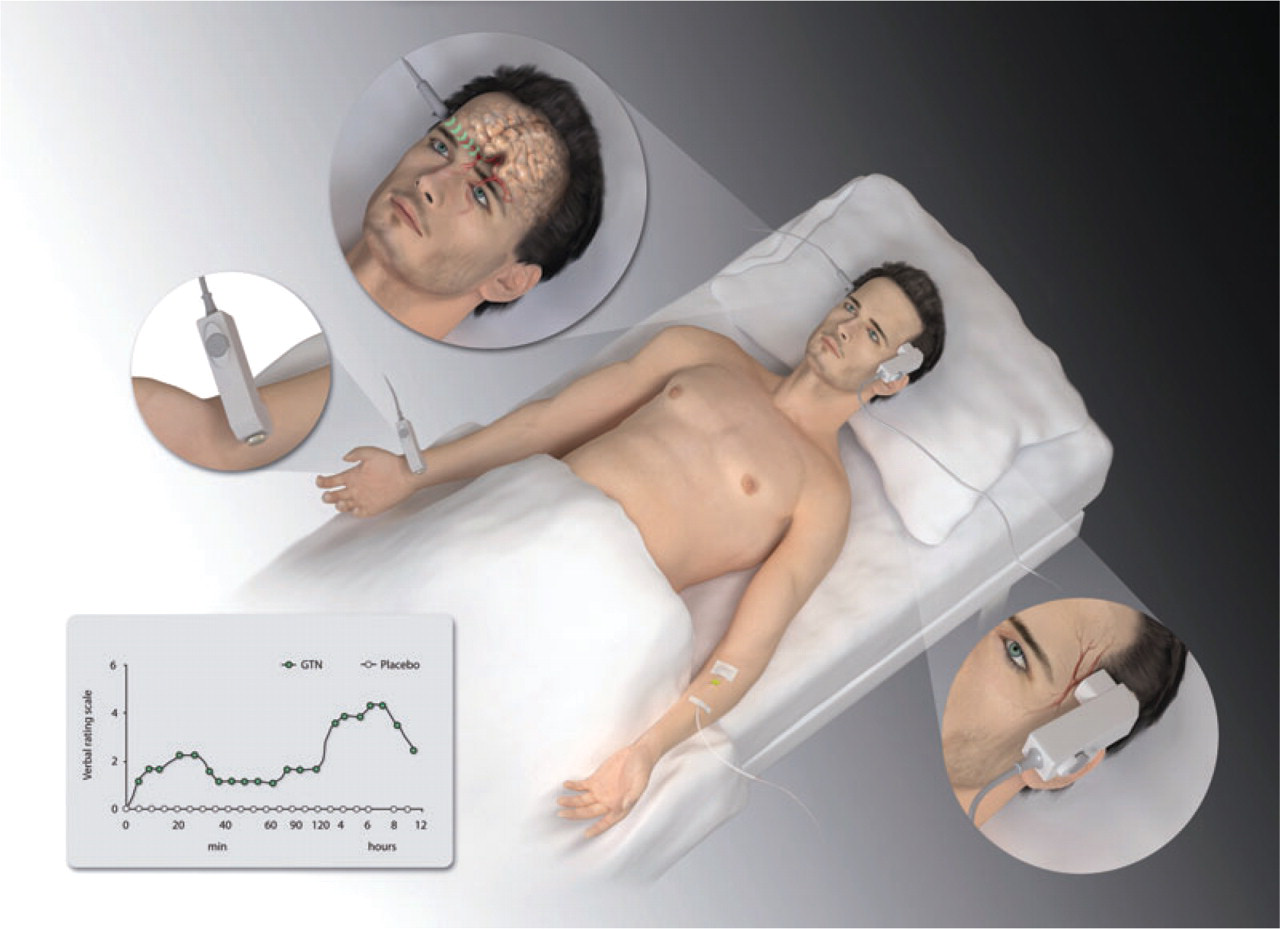
Human provocation model.
Box 2
In the main version of this model patients with migraine are randomly allocated to receive intravenous infusion (25 min) of ‘target substance’ or placebo (isotonic saline) in a double-blind, crossover design. Headache intensity is recorded on a verbal rating scale from 0 to 10 (0, no headache; 1, a very mild headache (including a feeling of pressing or throbbing); 5, moderate headache; 10, worst imaginable headache) (94). The following haemodynamic variables are recorded at intervals: mean velocity of blood flow in the middle cerebral artery (VmeanMCA) by transcranial Doppler with hand-held 2-MHz probes (Multidop X; DWL, Sipplingen, Germany) (75); diameter of the frontal branch of the superficial temporal artery by a high-resolution ultrasonography unit (Dermascan C; Cortex Technology, Hadsund, Denmark: 20 MHz, bandwidth 15 MHz) (116). Heart rate and blood pressure are measured continuously throughout the study. The subjects are asked to complete a headache diary every hour until 10 h after discharge. The diary included headache characteristics and accompanying symptoms necessary to classify migraine (117). The glyceryl trinitrate (GTN) experimental model of migraine showed that in migraine patients infusion of GTN causes an immediate headache during the infusion and a delayed headache that is maximal around 6–7 h after the infusion (75). This delayed headache has the characteristics of typical attacks of migraine without aura.
We have used this model to validate NOS inhibition and CGRP receptor antagonism as antimigraine targets as discussed above. Furthermore, we have suggested from human experimentation that inhibition of cyclic adenylyl monophosphate (101) or cyclic guanylyl monophosphate (102) may be effective. Adenosine antagonism and vasoactive intestinal peptide seem a less likely principle according to our provocation studies (95, 97, 103). We are currently studying a number of endogenous ligands for their migraine-provoking action.
If human study is not possible, animal models should be used (Fig. 3). They are unfortunately not well validated. Nevertheless, the neurogenic inflammation model, the vascular stimulation model, the closed cranial window model and the dura sensitization model can contribute to a better understanding of the mechanisms of action of a new compound. Such studies may also increase the likelihood that a potential target is valid. If substances or targets seem promising in these models, the necessary toxicology should be done to allow human provocation experiments in order to increase the likelihood that a target is useful.
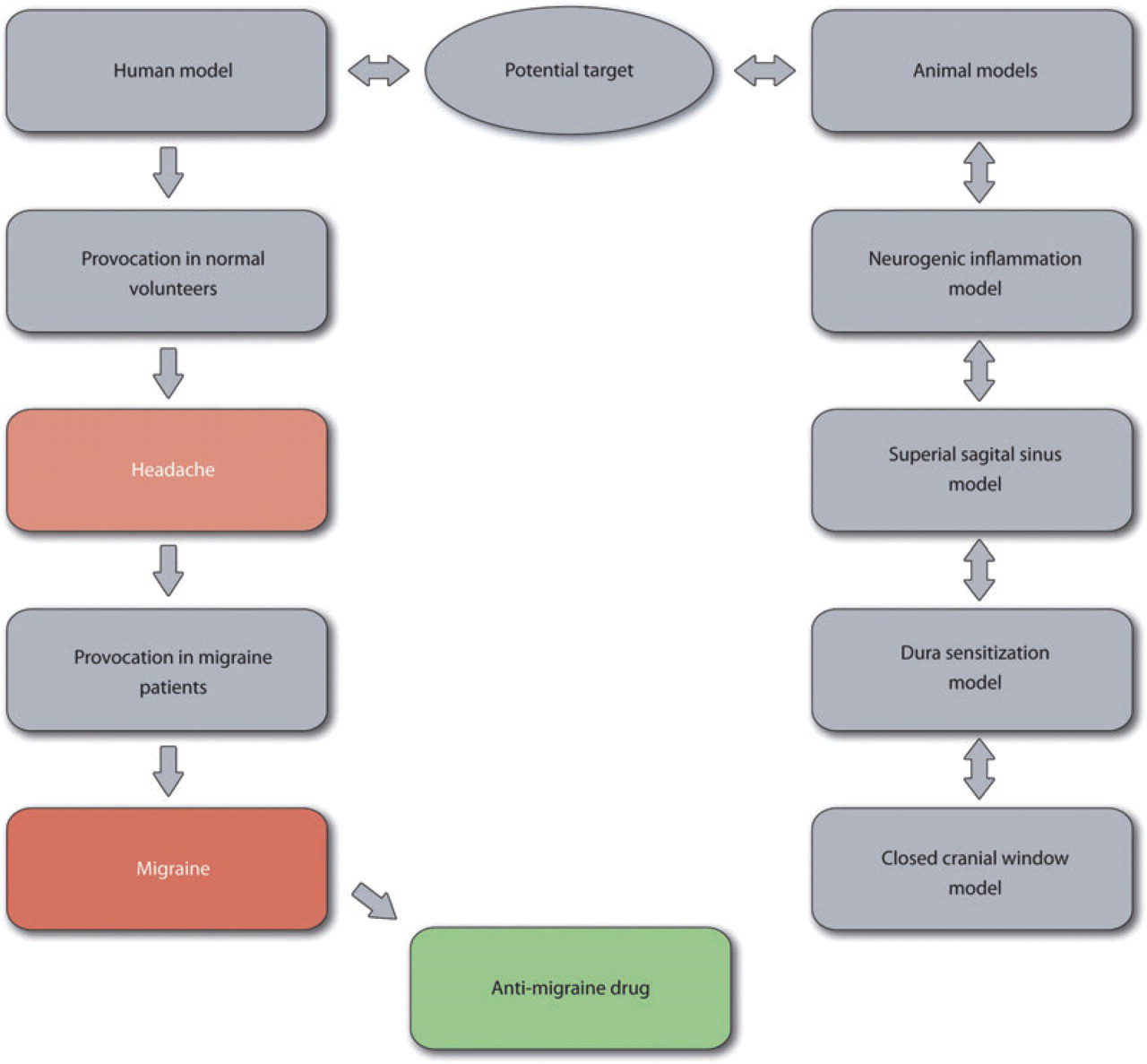
Proposed target identification for antimigraine drug. Only substances that produce substantial headache in healthy volunteers have the potential to induce migraine in migraine sufferers and only such substances should therefore be tested in migraine sufferers. If human study is not possible, animal models should be used to test substances or targets and their toxicology. This would allow human provocation experiments to increase the likelihood that a target is useful.
Another use for animal models is to study substances or mechanisms already known to induce migraine to better understand their mechanism of action. Thus, delayed migraine after nitroglycerin in humans had an animal correlate in activation of iNOS at 2–4 h after a nitroglycerin infusion (104). CSD, first validated in human studies, has been explored in animal studies demonstrating activation of matrix metalloproteinases (MMPs) that open the blood–brain barrier (105) and thus make it possible for the biochemical changes of CSD to act on nociceptors of the dura mater. This provides at least one mechanism whereby CSD and aura may cause headache, and MMPs may therefore represent a new target for migraine drugs.
Concluding remarks
The absence of validated screening models at first glance makes migraine a difficult disease for novel drug development. Nevertheless, there are more new interesting targets in migraine than in most other central nervous system (CNS) disorders. How is this possible? The answer lies in extensive use of human experimentation. Unlike almost all other CNS diseases it is possible to test naturally occurring substances and drugs for their headache- and migraine-provoking ability in human projects. If a naturally occurring substance can provoke migraine in human patients, then it is likely, although not certain, that blocking its effect will be effective in the treatment of acute migraine attacks. How should such a target be further pursued? If the offending molecule is a ligand, the corresponding receptor may be used for high-throughput screening of antagonists. The same is true if it is an ion channel opener. If it is an enzyme, enzymatic assays may be used. In other words, once a substance is known to provoke migraine, then high-throughput screening is easy. Usually the industry requests validation in several animal models before committing to the high expense of human toxicology. However, in migraine such models have uncertain validity. We therefore suggest conducting drug development based exclusively on human experimental evidence and knowledge of the location and function of the relevant receptor/ion channel/enzyme. This was the approach taken in developing the first CGRP antagonist and the first inhibitor of CSD, but it is such an unusual procedure for drug development that few companies accept it.