
Abstract
A pharmacological model of migraine is described using ultrasound vocalization (USV) of rats following central inflammation-induced sensitization to tactile stimulation. Central inflammation induced by intracerebroventricular injection of lipopolysaccharide (LPS) increased USV induced by an air current focused on the head and this was abolished by morphine and ketorolac, suggesting a nocice-ptive component. USV in naive rats were unaffected. Diazepam reduced USV in both inflamed and naive rats. The triptans, zolmitriptan and sumatriptan, both reduced USV in inflamed but not in naive rats, as did dihydroergotamine, and the calcitonin gene-related peptide (CGRP) antagonists αCGRP(8-37) and BIBN4096BS. The neurokinin-1 antagonist L-733-060 had no effect in either inflamed or naive rats when given after induction of inflammation, but when given with the LPS it prevented the augmentation of USV. This profile of activity of agents proven to be effective in the clinic suggests this model can be used to predict novel therapeutic agents for migraine.
Introduction
Management of migraine pain currently fall under two categories: pain-relieving (abortive) and preventive medication, both of which have limited efficacy and can be associated with complications. Development of new therapies for migraine has been hampered by the paucity of animal models for migraine. Moskowitz et al. (1) introduced an animal model of neurogenic dural inflammation. Electrical stimulation of the trigeminal ganglion resulted in the release of inflammatory neuropeptides from primary sensory afferents in the dura, triggering endothelial activation, vasodilation, mast cell degranulation and plasma protein extravasation (PPE). This model greatly improved our understanding of the pathophysiology of migraine and has been widely used to characterize many actual antimigraine drugs such as sumatriptan, aspirin and dihydroergotamine in vivo (2–4). However, it is not considered to be predictive of new antimigraine therapies. For example, pharmacological agents that inhibit PPE in the rat neurogenic dural inflammation model such as neurokinin (NK) 1 antagonists, valproic acid and bosentan have, subsequently, been shown to be ineffective in migraine (5–8). The availability of good therapeutically predictive animal models for migraine would be of great advantage in evaluating better treatments and understanding the pathophysiology and pathogenesis of migraine.
Extensive studies in both animals and man have led to the current view that a key component of the initiation of migraine is the release of large amounts of potassium and hydrogen ions, as proposed to occur during the phenomenon of cortical spreading depression (9–12). This is accompanied by the release of various neuroactive factors, such as nitric oxide (NO), adenosine and arachidonic acid, into the extracellular and perivascular space causing vasodilation in the cortex, pial vessels and dura mater. These molecules can activate and sensitize perivascular trigeminal afferents supplying the dura mater. Activation of meningeal nociceptors, in turn, release neuropeptides, including substance P and calcitonin gene-related peptide (CGRP) (13, 14), leading to neurogenic inflammation in the dura mater and subsequent further release of inflammatory mediators such as bradykinin, histamine, prostaglandins and serotonin (1, 2). This reinforcing cycle of release of neuronal sensitizing agents results in meningeal primary afferent neurons becoming mechanically hypersensitive. It is this intracranial mechanical hypersensitivity that may contribute to the pain of migraine headaches. Additionally, central sensitization occurs as a result of continued stimulation of these nociceptive fibres on second-order neurons. Increases in central trigeminal neuronal responses to the converging inputs originating from meningeal and cutaneous afferents (13, 15, 16) may be responsible for the increased response in migraineurs to previously innocuous stimuli (heat, cold and mechanical stimulation) applied to the face and scalp.
Cutaneous allodynia has also been described following application of chemical irritants to rat dura (13). Chemical or electrical stimulation of the dura mater resulted in decreased mechanical (air-jet, brush, pressure) and thermal (heat, cool) thresholds and increased facial cutaneous receptive fields (vibrissal pad, lower face, nose, eye) (17, 18).
There are a few reports of pain-related behavioural responses following inflammation induced in the dura or brain. Malick et al. (19) have demonstrated that inflammation of the dura of rats, induced by the administration of an ‘inflammatory soup’ to the dura in conscious animals, suppressed food intake, as seen in migraine patients. More directly related to nociceptive thresholds, Oshinsky and Gomonchareonsiri (20) have shown that multiple infusions of a similar ‘inflammatory soup’ in rats induced a facial mechanical allodynia, as measured by withdrawal thresholds to von Frey stimulation. We considered ultrasonic vocalization (USV) as an alternative to a withdrawal reflex response. USV is a primitive form of communication, with an average frequency of 22 kHz in rats, and has been suggested to relate to the emotional and/or motivational state of the animal (21–24). USV is evoked by a wide variety of factors such as distress/anxiety/fear (fights, predator, electric shock, air puff, acoustic startle, separation from litter), nociception, cold stress, withdrawal from drug abuse, and certain social interactions (21, 22, 25, 26). There are several reports relating USV to nociception with a correlation between intensity of nociception and degree of vocalization as well as modulation of USV by analgesics (27–29).
A common feature of these mechanistic studies on trigeminal nociceptive mechanisms is the induction of an inflammatory sensitization of trigeminal afferents using a variety of means, e.g. inflammatory soups, electrical stimulation, etc. A powerful inflammatory stimulus is the endotoxin lipopolysaccharide (LPS), which has been used extensively to induce systemic and central nervous system inflammatory responses. LPS injected into the brain parenchyma or ventricular system has been shown to induce an extensive inflammatory response throughout the ventricular system and the subarachnoid space (30, 31). Activation of microglia cells and astrolysis occurs in the temporal lobe and limbic system, with recruitment of neutrophils and monocytes. The inflammatory response is extensive, with neutrophils and macrophages abundant in the ventricular space and adjacent to ependymal or choroid epithelial surfaces as well as inflammatory exudate evident in the subarachnoid space.
As these meninges are innervated by trigeminal sensory afferents, we chose the intraventricular route to achieve maximal inflammatory sensitization of the trigeminal system.
With this in mind, the intention was not to develop a disease model for migraine but, rather, to ensure robust sensitization of the trigeminal system, centrally and then use USV as a behavioural end-point indicative of hypersensitivity. In the present study, our goals were threefold. First, we evaluated USV as an end-point for measuring LPS-induced cerebral inflammatory hyperalgesia. Second, we determined if pharmacological agents known for their analgesic properties could modulate USV. Lastly, we evaluated LPS-induced USV as a novel assay to assess clinical efficacy of migraine therapies.
Materials and methods
Subjects and housing
Male Sprague Dawley rats (175–200 g, Charles River, St Constant, Canada) were housed in groups of five in a temperature-controlled room (21°C, 40–70% humidity, 12 h light–dark) and were acclimatized in the animal facility for at least 1 day prior to use. The AstraZeneca Animal Care Committee approved all experimental protocols. Experiments were performed during the light phase of the cycle. Animals had food and water ad libitum and were sacrificed immediately after data acquisition.
Sample
Experiments included rats with and without intracerebroventricular (ICV) injections of LPS. Typically, in the LPS studies, four of the five groups were injected with LPS, one of the four groups served as the vehicle control and the other received the drug treatment. Experiments investigating any effects on naive rats were conducted in animals that had not been injected with LPS.
Administration of LPS
Rats were habituated in the experimental laboratory for 15–20 min prior to any manipulation. Central inflammation was induced by ICV administration, into the lateral ventricle, of 2.4 µg of LPS (endotoxin of Gram-negative Escherichia coli bacteria serotype 0111:B4; Sigma, St Louis, MO, USA) in a volume of 10 µl, using standard stereotaxic surgical techniques under isoflurane anaesthesia. The skin between the ears was pushed rostrally and a longitudinal incision of about 1 cm was made to expose the skull surface. The coordinates determined the puncture site: 0.8 mm posterior to the bregma, 1.5 mm lateral (left) to the lambda (sagittal suture) and 5 mm below the surface of the skull (vertical) in the lateral ventricle. LPS was injected via a sterile stainless steel needle (26G, 3/8 in) attached to a 100-µl Hamilton syringe. Following injection of LPS, the needle remained in place for an additional 10 s to allow diffusion of the agent. After removal of the needle, the incision was closed and the rat was returned to its original cage and allowed to rest for a minimum of 3.5 h prior to testing.
Experimental set-up for induction of USV
The rats remained in the experimental laboratory following LPS injection and drug administration until shortly before testing for USV, when they were moved to an anteroom to the laboratory. Subsequently, one rat at a time was brought into the testing laboratory and placed in a clear Perspex box (9 × 9 × 18 cm), which was then placed in a sound-attenuating ventilated cubicle measuring 62(w) × 35(d) × 46(h) cm (BRS/LVE; Div. Tech-Serv Inc. Beltsville, MD, USA). USV was evoked by blowing air into the box from a nozzle at one end of the Perspex box. The output nozzle for the air jet was 0.32 cm in diameter (AirStim; San Diego Instruments, San Diego, CA, USA) and each air jet lasted 0.25 s, with an intensity of 517 kPa and a frequency of 0.1 Hz (1 jet/10 s). A maximum of 10 jets was administered, or until vocalization started, whichever came first. The first air jet marked the start of recording. USV was recorded for 10 min using microphones (G.R.A.S. sound and vibrations, Vedbaek, Denmark) placed inside each cubicle. Frequencies between 0 and 32 kHz were recorded, saved and analysed using LMS equipment and software (LMS CADA-X 3.5C, Data Acquisition Monitor, Time Data Processing Monitor and UPA (User Programming and Analysis); LMS, Troy, MI, USA).
Experimental set-up for assessment of cold allodynia
Rats were shaved, either the back or periorbital area of about 1 cm2, under anaesthesia 1 day prior to any other manipulation. They were then injected with LPS as mentioned above. Testing was done 3.5 h later, and any drug administration followed the standard procedure and was given 30–60 min prior to testing (see Drugs section).
To test the response to a thermal stimulus (set at 0°C), rats were placed on a plate floor and hand-restrained while the thermal sensory device (TSA-2001 Thermal Sensory Analyser, thermode, diameter 5 mm; Medoc, Durham, NC, USA) was placed in contact with the shaved area and the withdrawal latency was measured. The first withdrawal response was considered to be due to the physical restraint therefore the second withdrawal was taken as the response to the stimulus.
Drugs
All drugs were pH-adjusted between 6.5 and 7.5 and administered in a volume of 4 ml/kg systemically or 20 µl/rat centrally. Morphine (Sigma) was dissolved in saline and administered by subcutaneous (s.c.) injection 30 min prior to testing at doses of 2.5–5.0–10.0 µmol/kg. Naloxone (RBI, Natick, MA, USA) was dissolved in saline and administered by s.c. injection 40 min prior to testing at a dose of 2.0 µmol/kg. Diazepam (Sabex, Boucherville, QC, Canada) was dissolved in saline and administered by s.c. administration 30 min prior to testing at doses of 1.05–3.5–5.3 µmol/kg (0.3–1.0–1.5 mg/kg). Ketorolac (Sigma) was dissolved in saline and administered per os (p.o.) 60 min prior to testing at doses of 53–106–159 µmol/kg (20–40–60 mg/kg). Zolmitriptan (AstraZeneca) was dissolved in 10% 2-hydroxypropyl-β-cyclodextrin (HBC) and administered by s.c. injection 30 min prior to testing at doses of 4.3–8.7–17.4 µmol/kg (1.25–2.5–5 mg/kg). Sumatriptan (Imitrex, local pharmacy) was dissolved in saline and administered by s.c. injection 30 min prior to testing at doses of 50–100–200 µmol/kg (15–30–60 mg/kg). Dihydroergotamine (Sigma) was dissolved in 10% HBC and administered p.o. 60 min prior to testing at doses of 1.9–3.8–7.6 µmol/kg (2.5–5–10 mg/kg). Human αCGRP(8–37) (Sigma) was dissolved in 0.25% dimethylsulphoxide (DMSO) and administered by ICV injection 30 min or 3.5 h (co-injection with LPS) prior to testing at doses of 12.5–25–50 nmol/rat. BIBN4096BS (synthesized in-house) was dissolved in saline and administered by ICV injection 30 min prior to testing at doses of 2–20–200 pmol/rat. N(G)-monoethyl-L-arginine (L-NMMA; Sigma) was dissolved in saline and administered by s.c. injection 20 min prior to testing at doses of 50–100–150 µmol/kg. L-733-060 (NK-1 antagonist; Sigma) was dissolved in 0.25% DMSO and administered by ICV injection 30 min or 3.5 h (co-injection with LPS) prior to testing at doses of 2–20–200 nmol/rat. L-733-061 (inactive enantiomer NK-1 antagonist; Sigma) was dissolved in 0.25% DMSO and administered by ICV injection 3.5 h (co-injection with LPS) prior to testing at doses of 2–20–200 nmol/rat. Following drug administration, animals were returned to their original cages until time of testing.
Analysis
The USV recordings were filtered (between 20–24 kHz) and run through a series of statistical and fourier analyses to calculate the parameters of interest. All data are expressed as the mean ± standard error of mean (
Results
USVs in naive and LPS-treated rats
LPS administration led to the development of a clear central inflammatory response, as evidenced by a 20–60-fold increase in brain levels of interleukin (IL)-6, IL-1β, tumour necrosis factor-alpha, as well as an increase in macrophage marker, ED-1 (unpublished observation). LPS-treated rats showed behavioural signs of a generalized ‘illness’ response. These symptoms included immobilization, visible signs of discomfort (posture, piloerection, squint-eyed), loss of appetite, reduced startle threshold (phonophobia) and a reduced escape reflex with little or no aggression.
A series of air jets of increasing intensity, but constant duration (0.25 s) and frequency (0.1 Hz) was applied to the face of the rats. Naive rats emitted a stable, constant level of USVs at all air intensities tested, varying between 88 and 106 USVs (Fig. 1a). In contrast, a highly significant intensity-dependent number of USVs was measured in LPS-treated rats, increasing from 75 USVs at 69 kPa to 341 USVs at 517 kPa (n = 8,
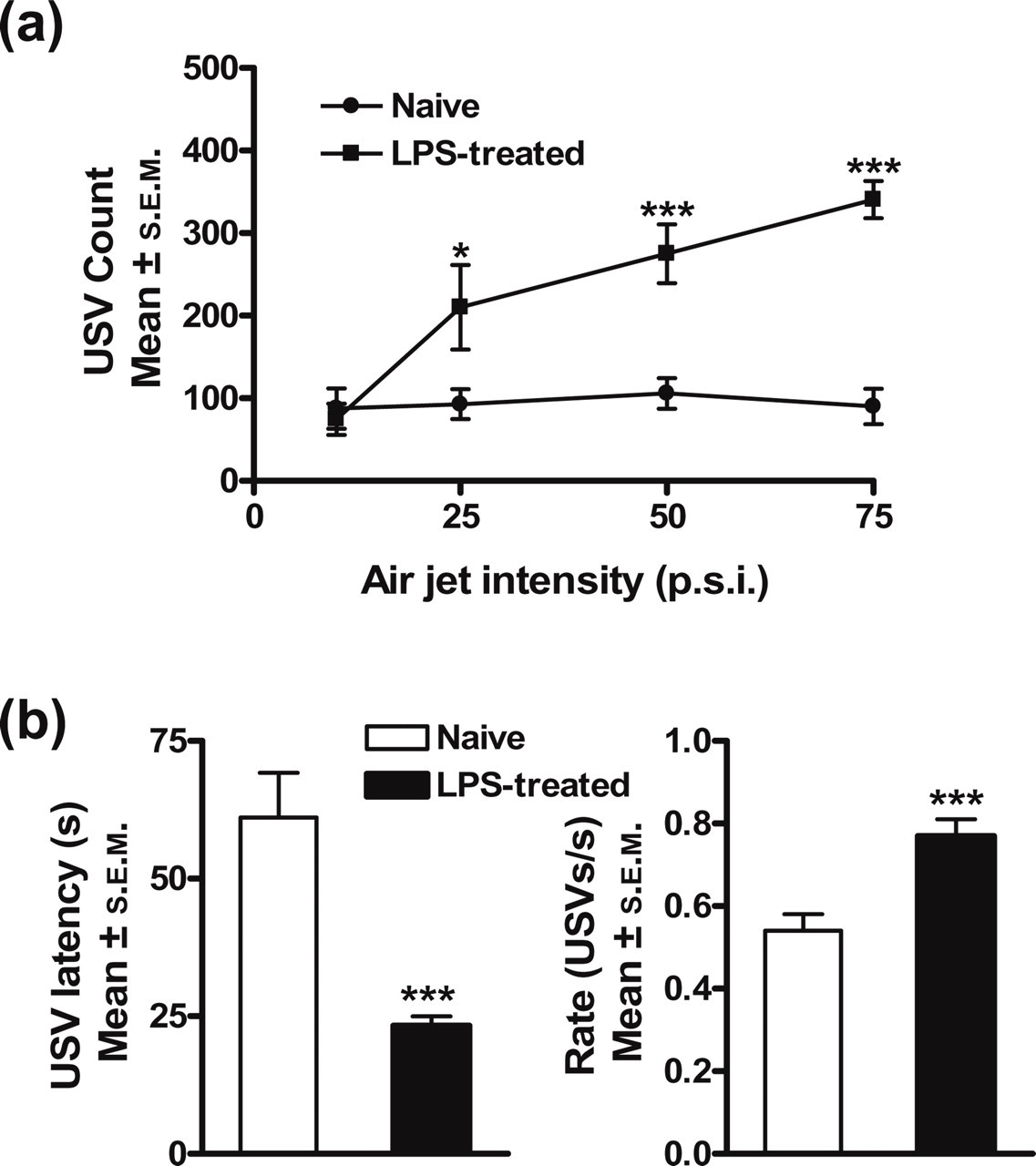
Air jet-induced ultrasound vocalization (USV). (a) Mean (±
For all subsequent pharmacological experiments, 517 kPa was chosen as the stimulus intensity, since a robust and consistent number of USVs can be evoked in LPS-treated rats (pooled data n = 92, t-test P < 0.00001). A cut-off of 10 air jets was selected, as the average number of air jets required to evoke a response was eight for naive and four for LPS-treated rats. Furthermore, the latency to vocalize and rate of vocalization were also measured. The latency decreased threefold, from 61.1 ± 8.1 s in the naive, uninflamed group to 23.3 ± 1.7 s in the LPS-treated group (pooled data n = 92, t-test P < 0.00003, Fig. 1b), and the instantaneous rate increased from 0.54 ± 0.03 USVs/s to 0.77 ± 0.04 USVs/s, respectively (pooled data n = 92, t-test P < 0.00004).
The presence of cold allodynia in LPS-inflamed rats
In Fig. 2, the periorbital region of naive rats was more sensitive to cold stimulation (0°C) when compared with the back of the animal. In rats that received ICV administration of LPS, there was an increase in cold sensitivity in the periorbital region (n = 8, t-test P = 0.0008), whereas no change was noted on the back of the animals (n = 8, t-test P = 0.29).

Lipopolysaccharide (LPS)-induced cold allodynia. Mean (±
Effects of opioids, diazepam and non-steroidal anti-inflammatory drugs
Morphine (2.5–10 µmol/kg) dose-dependently reduced USVs in the LPS-treated rats (Fig. 3a, n = 8,

Morphine reverses lipopolysaccharide (LPS)-induced ultrasound vocalization (USV) via the µ opioid receptor. (a) Mean (±

Diazepam reduces ultrasound vocalization (USV) in all rats. Mean (±
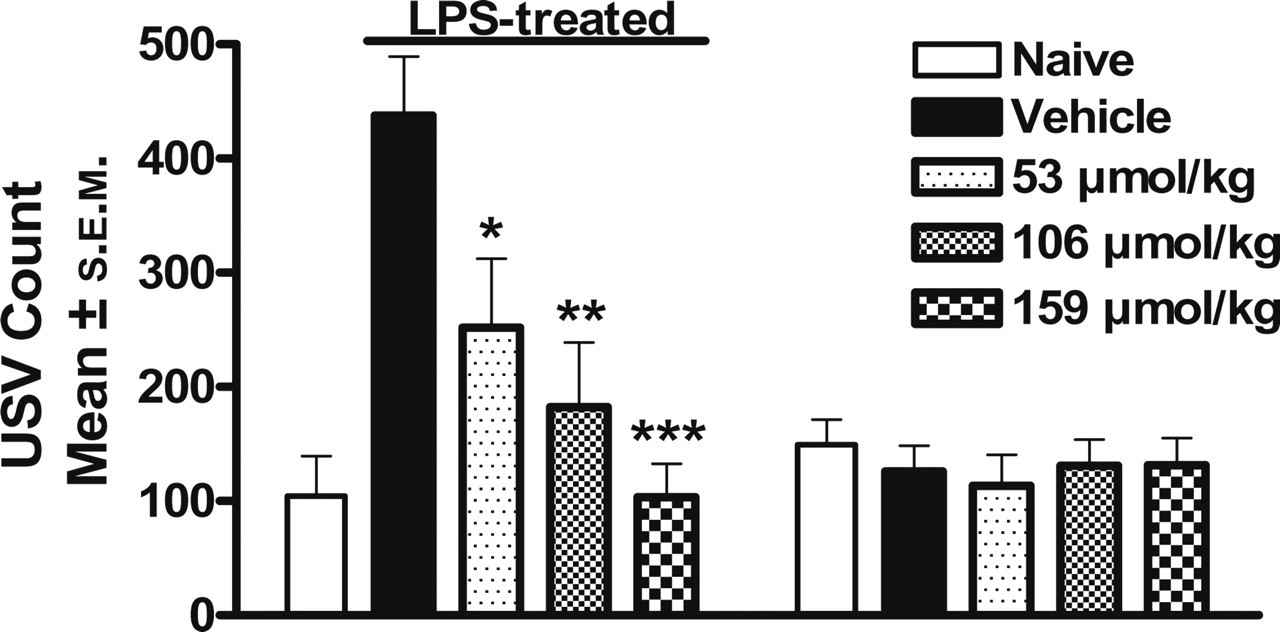
Non-steroidal anti-inflammatory drug reduces ultrasound vocalization (USV) in lipopolysaccharide (LPS)-treated rats. Mean (±
Clinically effective migraine drugs
Zolmitriptan (4.3–17.4 µmol/kg, s.c.) given 3 h after LPS injection reduced USV in LPS-treated rats (n = 8,
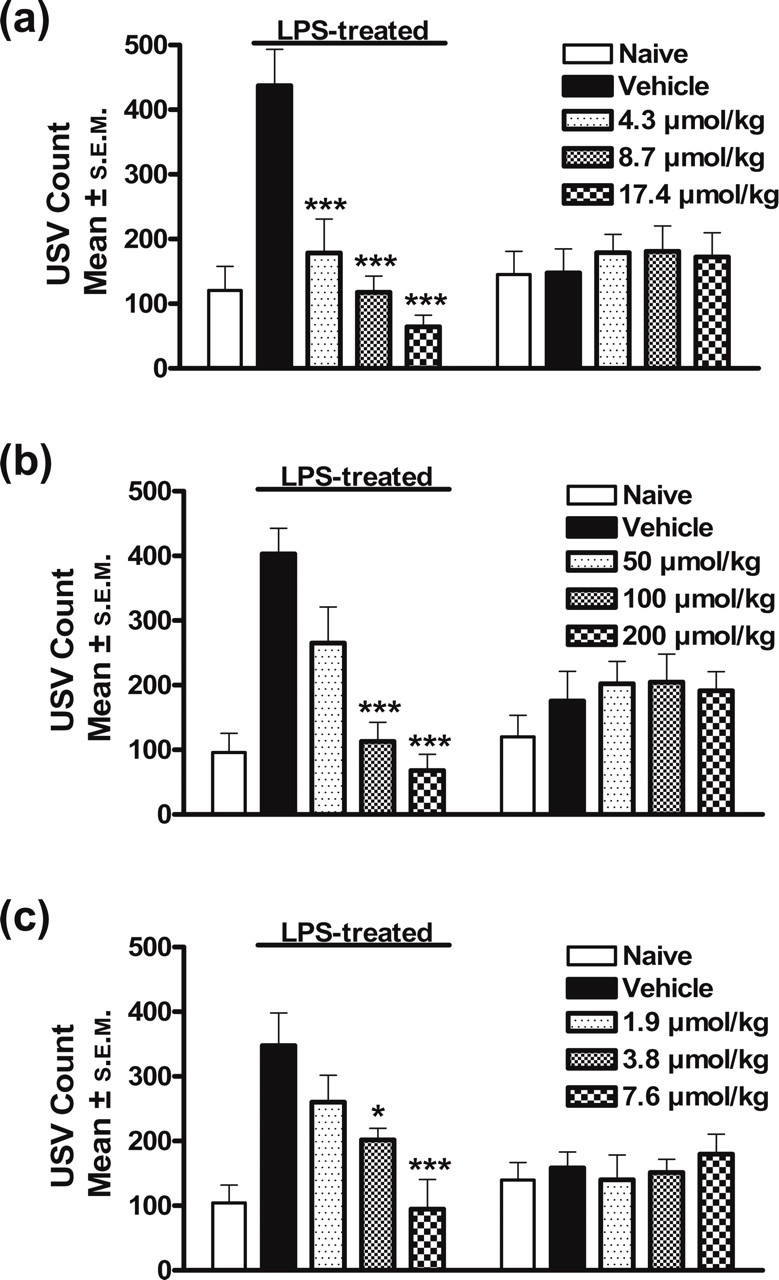
5HT1B/1D receptor agonists reduce ultrasound vocalization (USV) in lipopolysaccharide (LPS)-treated rats. Mean (±
Reversal of cold allodynia by ketorolac and zolmitriptan
Ketorolac (159 µmol/kg, p.o.) completely reversed LPS-induced cold allodynia in the periorbital region to levels seen before LPS injection (Fig. 7, n = 8,

Ketorolac and zolmitriptan reverse cold allodynia in lipopolysaccharide (LPS)-treated rats. Mean (±
Potential migraine therapeutic agents
Human αCGRP(8–37) administered (12.5–50 nmol/rat, ICV) 3 h after LPS injection, or co-administered with LPS, dose-dependently reduced USVs in inflamed rats (Fig. 8a, n = 8,

hCGRP(8–37) and BIBN4096BS reduce ultrasound vocalization (USV) in lipopolysaccharide (LPS)-treated rats. Mean (±

N(G)-monoethyl-L-arginine (L-NMMA) reduces ultrasound vocalization (USV) in lipopolysaccharide (LPS)-treated rats. Mean (±
NK-1 antagonist
The selective NK-1 antagonist, L-733-060 (2–200 nmol/rat, ICV) had no effect on USV (Fig. 10) when administered 3 h after the inflammatory response had become established (n = 8,
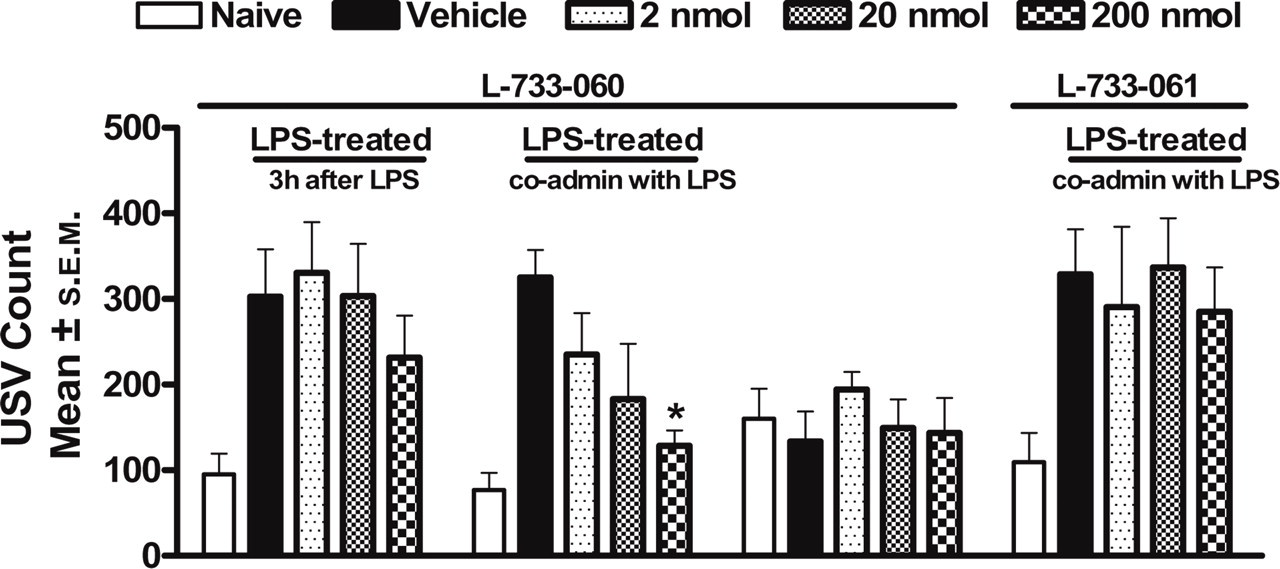
Neurokinin-1 antagonist effect in ultrasound vocalization (USV). Mean (±
Discussion
We have developed a pharmacological model of migraine in vivo by inducing an inflammatory response in, and sensitization of, the trigeminal system via central administration of LPS, an E. coli bacterial cell membrane glycolipid well-characterized for its proinflammatory activity (32–35). Airflow was chosen as the stimulus, being a natural, non-noxious mechanical stimulus that has been shown to evoke reliable and consistent USV emission in rats (36). Electrophysiological studies in rats have clearly shown that there is sensitization of the trigeminal nociceptive pathway to non-noxious tactile stimulation of the vibrissae following inflammation of the dura (17, 18). The increase in USV seen in the present model following induction of inflammation may reflect a behavioural correlate of this neural substrate sensitization. Clearly, there was an increase in USV in response to the airflow directed towards the head following induction of cerebral inflammation. When the air jet was disconnected but the same procedure followed, there was no USV response and as, in this situation, the noise remained the same, it is clear it was the air movement and not the accompanying noise that induced the response. However, although the jet of air was directed towards the face, it was not possible to conclude that it was movement of the vibrissae, exclusively, that gave rise to the observed USVs, although in view of the literature showing enhanced neuronal responses to vibrissae movement after central inflammation, this is the most likely explanation.
The hypersensitivity does, however, appear to be a trigeminally mediated phenomenon, as an enhanced aversive response to cold after LPS injection was observed on the face, but not on the back of the animal. The prevention of this response by triptans is further support for the cold response being mediated via the trigeminal system.
USV has been associated with many different states in an animal, ranging from social interactions to fear/anxiety and pain (21–29). There is not an extensive literature relating USV to pain, and the conclusions regarding a causal link between pain and USV are mixed, with some studies supporting USV as a measure of nociception (37, 38) and others finding no link (39, 40). It is not clear why there are such marked differences, but it may reflect the type of nociceptive insult, the type of stimulus, as well as the test setting or environment.
Kemper et al. (41) have explored the potentiating effect of LPS on a nocifensor response to an intracranial infusion of capsaicin. They, like ourselves, observed a reduction in motility due to LPS itself and potentiation of some of the behavioural responses to, presumably, trigeminal afferent stimulation by capsaicin. They did not, however, record any externally evoked nocifensor behaviours.
In this model there are two quite distinct pharmacological USV profiles. Anti-nociceptive agents such as NSAIDs or morphine do not affect USV induced in the naive rat, which suggests that this aspect of the response is not nociceptive in nature. However, following inflammation there is an increase in USV and this increased, or sensitized, response is reduced by NSAIDs and morphine, suggesting a nociceptive component. Similarly, the exaggerated response to cold was also blocked by an NSAID. The response to morphine in naive rats at the highest dose of morphine tested is almost certainly indicative of a sedative effect of morphine at this dose. The reduction of USV in both inflamed and naive rats by diazepam at doses that were clearly sedative supports this interpretation.
What is particularly interesting is the profile of response to proven clinically effective and putative antimigraine therapeutic agents. There is a consistent pharmacological profile of response, with such agents profoundly reducing and even abolishing the increase in USV induced by inflammation, but with no effect on the USV responses in naive rats.
The most common abortive treatment for migraine is with triptans, and the reduction in LPS-induced USV is seen with both zolmitriptan and sumatriptan, with zolmitriptan being more potent, as is the case in the clinic (42). This pattern was also seen with dihydroergotamine, which has been used, clinically, to treat migraine (43). ‘Triptans’ are 5HT1B/1D receptor agonists and, amongst other actions, have been shown to reduce neuronal responses in the trigeminal nucleus to convergent stimuli from the dura and vibrissa (17) and, as referred to above, trigeminal neuronal responses to dual dural and facial inputs are sensitized when inflammatory mediators are applied to the dura (18). In addition, naratriptan can reduce noxious electrically evoked responses in the trigeminal neurons, but not in the spinal cord, suggesting 5HT1B/1D receptors have a particular role in nociceptive processing within the trigeminal system (44).
Morphine also showed a differential inhibition of USV in the inflamed, but not the naive rat at a dose that was not, apparently, sedative. Use of narcotics in migraine is generally not recommended, but they are considered as a last resort for patients with moderate to severe pain who do not respond to non-opioid medication, or cannot tolerate other migraine medication. Mechanistically, morphine has been shown to block nociceptive neurotransmission within the trigeminal nucleus caudalis and inhibit neurogenic dural vasodilation. These effects are similar to what is seen with 5HT1B/1D agonists (45–47).
A similar pattern is seen with antagonists of CGRP. In this case, the peptide and non-peptide antagonists were given ICV. The peptide antagonist, hCGRP(8–37), was able to reduce USV when given either with the LPS or subsequently, suggesting a therapeutic potential for CGRP antagonists in prevention of migraine-induced pain. In man, CGRP given intravenously precipitates headaches (48) and a CGRP antagonist has been shown to be clinically effective in alleviating migraine (49). Mechanistically, CGRP may also have a role in sensitization of the trigeminal system, as it has been shown to facilitate vibrissal responses in the trigeminal nucleus and, interestingly, this facilitation can by blocked by a 5HT1B/1D agonist (17).
The iNOS inhibitor, L-NMMA, also showed an ‘antimigraine’ pharmacological profile. In man, NO metabolites, nitrites, were elevated in patients with migraine and cluster headaches (50), and a non-selective NOS inhibitor was effective in migraine in patients (51). There is also evidence linking NO mechanisms with the trigeminal system. Neurons in the trigeminal nucleus, which receive input from the dura, show an increase in activity when a NO donor is infused and the NOS inhibitor N(G)-nitro-L- arginine methyl ester decreases the spontaneous activity of such neurons (52, 53).
All these three mechanisms, 5HT1B/1D receptors, CGRP and NO, may well be interlinked. 5HT1D receptors are expressed on fibres containing CGRP, and it has been shown that NO can increase the release of CGRP and that this release is inhibited by sumatriptan (54, 55).
In contrast, the profile with the NK1 antagonist, L-733-060, is different, with no effect on LPS-induced USV when administered after the inflammation has developed. Clinically, NK1 antagonists have been shown to be ineffective in reducing migraine pain (6, 56). Intriguingly, however, there was a reduction of USV when the NKI antagonist was co-administered with the LPS. Mechanistically, this suggests involvement of Substance P in the early phases of inflammatory sensitization, but not once it is established. Only one NK1 antagonist has been evaluated in the clinic as prophylactic treatment for migraine, and it had no effect on frequency of migraine (57); however, there have been questions raised as to whether that NK1 antagonist had the optimal pharmacokinetic profile to test the concept (58). Within the context of this model and the trigeminal system, NK1 receptors and Substance P have been identified in the trigeminal system, but a centrally acting NK1 antagonist had no effect on electrically evoked activity of trigeminal neurons in the cat (59). Additionally, electrically evoked dural vessel vasodilation in the rat was blocked by a CGRP antagonist, but not by an NK1 antagonist (60). It thus seems that NK1 receptors do not play a significant role with respect to trigeminal activation or consequential vasodilation.
In conclusion, this is not, nor was it intended to be, a disease model of migraine, but may well be a predictive pharmacological model of migraine. The profile of an antimigraine agent in this model is one that reduces the increase in USV seen in the LPS-inflamed rat, but with no effect on USV in the naive rat. Agents that do not reduce the LPS-induced increase in USV would not be predicted to be effective in migraine. Reduction in USV in both the inflamed and non-inflamed animals is harder to interpret, although in this study that profile has been seen only when there is clear evidence of sedation.
In summary, this model of tactile-induced USV following cerebral inflammation appears to show an interesting pharmacology highly predictive of efficacy in migraine. Further studies would be needed to explore the mechanistic elements in this model and to correlate those with known mechanistic aspects in migraine.