
Abstract
We tested the idea that migraine triggers cause cortical activation, which disinhibits craniovascular sensation through the nucleus raphe magnus (NRM) and thus produces the headache of migraine. Stimulation of the dura mater and facial skin activated neurons in the NRM and the trigeminal nucleus. Stimulation of the NRM caused suppression of responses of trigeminal neurons to electrical and mechanical stimulation of the dura mater, but not of the skin. This suppression was antagonized by the iontophoretic application of the 5-HT1B/1D receptor antagonist GR127935 to trigeminal neurons. Migraine trigger factors were simulated by cortical spreading depression (CSD) and light flash. Activity of neurons in the NRM was inhibited by these stimuli. Multiple waves of CSD antagonized the inhibitory effect of NRM stimulation on responses of trigeminal neurons to dural mechanical stimulation but not to skin mechanical stimulation. The cortico-NRM-trigeminal neuraxis might provide a target for a more universally effective migraine prophylactic treatment.
Introduction
We have recently advanced a hypothesis about the mechanism of initiation of migraine headache by trigger factors (1). We theorized that different pathways arising from neurons in a variety of cortical regions, each itself activated by a different trigger, might converge on the brainstem and, by releasing dural sensation from inhibitory control, result in the common symptomatology of migraine headache. In summary, we hypothesize that every migraine attack probably has a perceptible or imperceptible trigger; that these triggers produce as their first effect an activation of cortical neurons; that this activation inhibits the drive of brainstem neurons of the intrinsic pain control system; and that this inhibition selectively disinhibits trigeminovascular sensation.
The cortex was long ago suggested to be a possible ‘culprit’ in the generation of migraine (2), although this idea lay dormant for a century. The brainstem became a focus of attention for migraine research about 20 years ago in several laboratories, including our own, and there is increasing evidence that a ‘perturbation’ of the brainstem may lead to migraine (3–6). Two nuclei have been implicated in this defect—the periaqueductal grey matter (PAG) (5) and the nucleus raphe magnus (NRM) (7). The NRM in particular has been described as fulfilling a ‘gain control’ function (8), which is the role we foresee for it in migraine. We have summarized evidence that such a neural pathway might exist (1) and here present experimental evidence that some components of it behave in a way consistent with the hypothesis. In this study, we have examined the possible involvement of a cortico-NRM-trigeminal nucleus neuraxis in the generation of migraine headache, but similar results have been obtained for the cortico-PAG-trigeminal nucleus neuraxis as well. Preliminary results were presented at the 2006 Migraine Trust International Symposium (1) and at IBRO2007 (9).
Methods, materials and protocols
The experiments described here used methods, materials and protocols which we have previously described in full (10–13). Experiments were conducted in 37 cats anaesthetized with α-chloralose. Cats were anaesthetized by induction with isoflurane (5% in a 30% oxygen in air mixture) followed by an intraperitoneal injection of α-chloralose (60 mg/kg). Polyethylene catheters were inserted into the femoral artery and vein to allow continuous measurement of blood pressure and heart rate and to administer drugs and fluids. Bilateral pneumothoraces were made and cats were intubated and ventilated with 30% oxygen in air to maintain end expiratory CO2 at 3.5–4%. Core body temperature was monitored by a rectal thermistor and maintained at 37–38°C by a servo-controlled heating blanket.
Animals were mounted in a stereotaxic frame and the lateral C1 spinous processes were secured to the frame with clamps to minimize brainstem movement during recording. The trigeminal nucleus and upper cervical spinal cord were exposed by a C1/C2 laminectomy and occipital craniotomy. The dura was reflected away from the spinal cord and part of the cerebellum was removed to expose the obex. Immediately before insertion of an electrode, the pia mater was carefully removed with titanium micro forceps.
Two rectangular craniotomies (approx 1.5 × 0.8 cm, long axis longitudinal, mid-point at A/P 5 mm, one either side of the midline) were carefully drilled under a saline drip to expose the underlying dura mater.
To allow access to the NRM, a midline rectangular craniotomy was made over the cerebellum by carefully grinding away portions of the parietal bones, exposing the bony tentorium. The tentorium, down to a depth of 6 mm, was carefully drilled away from the cerebellar side with a low-speed dental burr, under micropsic observation. Care was taken not to abrade the dura and this procedure was carried out under 1.5% supplementary isoflurane anaesthesia, to prevent the development of inadvertent cortical spreading depression (CSD) (14).
Multi-barrelled pipette electrodes with a central tungsten wire were used to record discharges of neurons in the trigeminal nucleus. Electrodes were positioned orthogonal to the surface, from 2 mm rostral to 4 mm caudal to the obex and 3.0–4.5 mm lateral to the midline, their tips on the dorsal surface of the brainstem. Bacteriological agar (2% in saline, 40°C) was poured onto the exposed brainstem and allowed to set to reduce disruption of recording caused by movement related to cardiac pulsations. A piezoelectric microdrive was used to advance the electrode into the brainstem 5 μm at a time, to a depth no greater than 3000 μm.
A Dagan 6400 iontophoresis generator provided the current for ejection and retention
of drugs from the electrode barrels. In every iontophoresis experiment, four of the
barrels of the electrode were filled with: saline solution (0.9% NaCl); 3
Single-barrelled tungsten-in-glass recording electrodes were used to record neurons in the NRM. They were lowered by hand to a point about 3 mm above the NRM and a piezoelectric microdrive used to advance them further into the brainstem 5 μm at a time, to a depth no greater than 2000 μm below the NRM. We looked for responsive neurons in the NRM proper (A/P =−9, L = ±1, H =−8.0) and in the rostral ventromedial medulla (RVM) in which the NRM is embedded (15).
Cutaneous receptive fields were classified according to standard criteria defined by Hu et al. (16) as nociceptive specific (NS: responding only to pinch with toothed forceps or deep pressure with a surgical probe), wide dynamic range (WDR: responding to pinch or deep pressure and light touch or brush with a small surgical probe) or low threshold mechanoreceptors (LTM: responding only to brush or light touch). The centre of each cutaneous receptive field was stimulated electrically with a pair of stimulus isolated stainless steel needle electrodes, which delivered supramaximal shocks (10–120 V, 250 μs).
We adapted a previously described device (17) so as to apply von Frey hairs to the dura or skin in a time-synchronized and repeatable manner and also used conventional sensory stimulation methods for probing receptive fields. A von Frey hair, two numbers above the threshold for the neuron under test, was mounted in the automated von Frey device and advanced with a micromanipulator until the tip of the hair lay just above the centre of the receptive field of the neuron. Repetitive or intermittent single von Frey stimuli were then applied by activating the device from a stimulator, which was in turn driven by the data-acquisition software. The effect was to substitute electrical stimulation with calibrated von Frey stimulation in the data acquisition protocols.
The exposed dura was explored with a range of von Frey hairs (number range 2.44–6.65). Receptive fields were classified as NS (responding only to number ≥ 5.07), WDR (responding to von Frey numbers 2.83–4.93) or LTM (responding to von Frey hair numbers < 2.83). The location, extent, modality and threshold von Frey hair number were recorded on drawings. The dura was stimulated electrically with chlorided bipolar silver ball electrodes which delivered stimulus-isolated, supramaximal single shocks (40–150 V, 0.4–1.5 mA) of 250 μs duration, every 3–5 s. One pole of the electrode lay at the caudal extremity of one craniotomy near the midline on the border of the superior sagittal sinus and the other in the same relative location in the opposite craniotomy (when recording in the NRM) or in the same craniotomy but more lateral to it (when recording in the trigeminal nucleus). For mechanical stimulation of the dura, a von Frey hair, two numbers above the threshold for the neuron under test, was mounted in the device described above and advanced under stereotaxic control until the tip of the hair lay just above the centre of the dural receptive field of the neuron under test. Repetitive or intermittent single von Frey stimuli were then applied by activating the device as for skin stimulation.
CSD was initiated with a 1-µl injection of 1
For experiments with light flash, the trigger output of a stimulator was passed inductively to the trigger input of a stroboscopic light source (18). The light output of the gun was directed into the eye, which was dilated by the topical application of atropine sulphate eyedrops. Flashes were delivered at 10/s, approximately half the critical flicker fusion frequency. Light flash continued until a maximum response had been achieved or for 10 min, whichever was the lesser time.
Stainless steel concentric bipolar electrodes (Rhodes, SNEX-200) were inserted into the NRM (coordinates as above). Trains of 1–5 square wave pulses of 50–200 μs duration and 100– 500 μA stimulus-isolated current at a frequency of 200–400 Hz were delivered at varying times before electrical or mechanical stimulation of the dura mater or skin.
Cortical neuronal activity monitoring via laser Doppler flowmetry
Blood flow was monitored with a pencil probe (P-431, TSI, diameter 0.15 cm), connected to a single-channel laser-Doppler perfusion monitor (Laserflo BPM403, Vasamedix, St Paul, MN, USA) (12, 19).
Statistical methods
To test for significance of changes in evoked responses and basal discharge
rates, Student's t-test, the critical ratio test
(20) and
Histology
Electrolytic lesions were made at the bottom of a ‘plunge’ by passing 5–10 μA d.c. cathodal current for 10–15 s through the recording electrode. At the end of each experiment the animal was deeply anaesthetized with pentobarbitone sodium and perfused with physiological saline followed by 10% formalin in phosphate-buffered saline. The brainstem and upper cervical spinal cord were removed and later cut at 50-μm sections on a freezing microtome and the sections stained with cresyl violet. Recording sites and the receptive fields associated with them were recorded on micrographs from the brain stem atlas of Berman (21).
Experiments were approved by the University of NSW Animal Care and Ethics Committee and conformed to its guidelines.
Protocols
Four series of experiments were carried out:
Effect of peripheral electrical, mechanical and light flash stimulation on neurons in the NRM.
Effect of CSD and light flash on the basal discharge rate of NRM neurons.
Effect of conditioning stimulation of the NRM on the responses of trigeminal neurons to electrical and mechanical stimulation of the skin and dura mater.
Effect of CSD on the NRM-stimulus-induced suppression of responses of trigeminal neurons to mechanical stimulation of the dura mater and skin.
Results
A. Effect of peripheral electrical, mechanical and single light flash stimulation on neurons in the NRM
Fourteen NRM neurons from eight cats were tested for responsiveness to peripheral stimulation. Most NRM neurons were spontaneously active with a discharge rate 1–20/s. NRM neurons responded to electrical stimulation of the dura and of the skin and to mechanical stimulation of the skin. Latencies and number of responses are shown in Table 1.
Characteristics of responsive neurons in the NRM

Responses of nucleus raphe magnus neurons to trigeminal electrical and mechanical (von Frey hair no. 4.56, solenoid-driven) stimulation of facial skin and electrical stimulation of dura mater. The latency of the response following mechanical stimulation includes that which arises from the inertia of the stimulating device.
B. Effect of CSD and repetitive light flash on the basal discharge rate of NRM neurons
Recordings were made from a total of 31 spontaneously active NRM neurons in 10 cats, under control conditions and during either CSD or light flash. Basal neuronal discharge rate was 9.6 ± 1.5/s (mean ± standard error, n = 41 observations), which is about twice the rate of NRM neurons in conscious cats (22). The discharge rate of 12 out of 17 neurons decreased significantly (critical ratio test, Student's t-test, Kolmogorov–Smirnoff test) following the initiation of CSD, falling from 9.7 ± 2.5 to 4.1 ± 1.5/s. The discharge rate reached a minimum at a time of 21 ± 13 min following the initiation of CSD. An example is shown in Fig. 1 (middle). At time 60 min after initiation of CSD, discharge rates had returned to 8.0 ± 2.6/s.
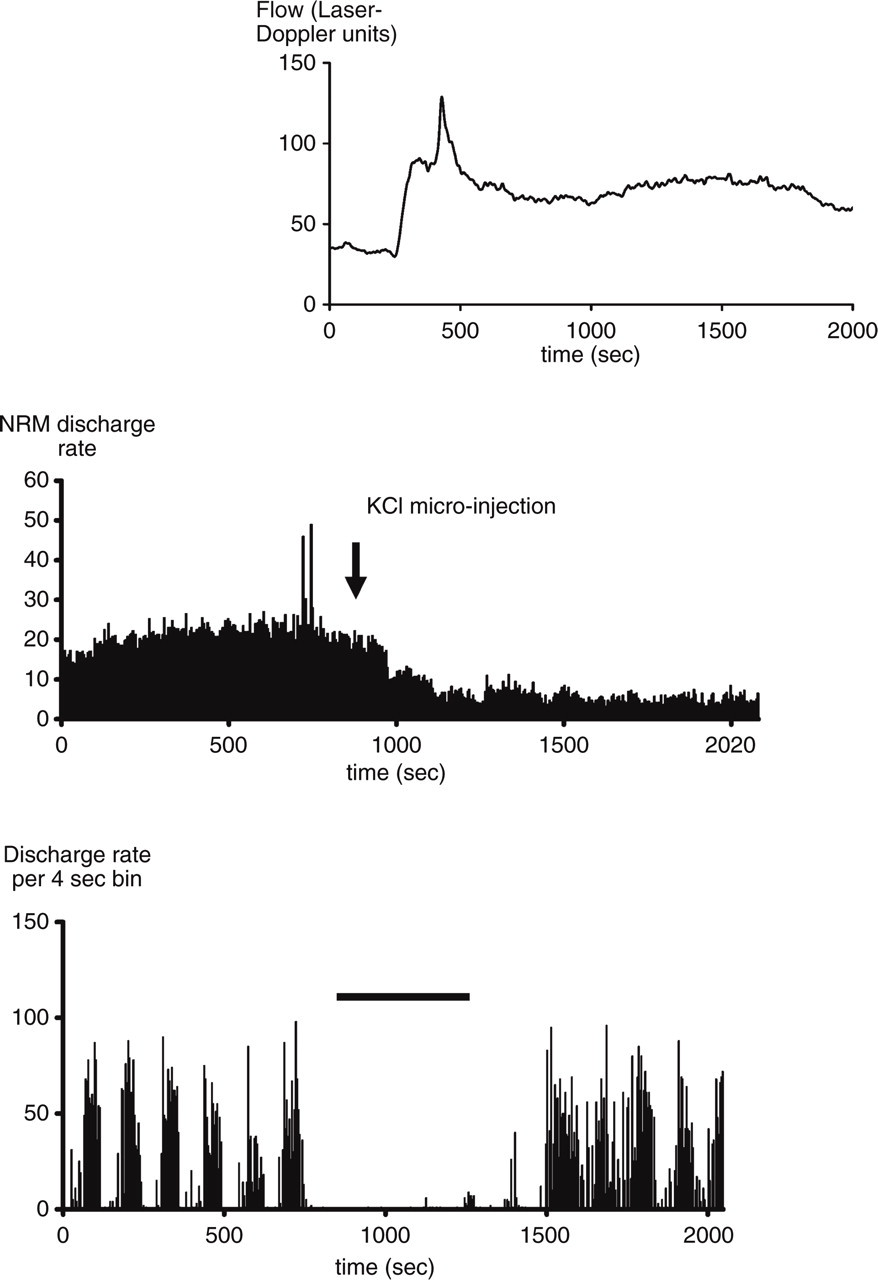
Examples from two neurons showing, top and middle: cortical spreading
depression (CSD) (indicated by top graph showing cortical flow increase)
decreases the baseline discharge rate of a nucleus raphe magnus (NRM)
neuron. One microlitre of 1
The mean discharge rate of 14 out of 26 neurons decreased following the initiation of repetitive light flash (critical ratio test), falling from 9.6 ± 2.0 to 5.8 ± 1.4/s. Discharge rate reached a minimum at 13 ± 5 min following the initiation of light flash. Discharge rates did not begin to return to normal until about 5 min after cessation of flash stimulation. At time 60 min after cessation of light flash, discharge rates had returned to 8.8 ± 2.6/s. An example is shown in Fig. 1 (lower).
C. Effect of conditioning stimulation of the NRM on the responses of trigeminal neurons to electrical and mechanical stimulation of the skin and dura mater
Recordings were made from 73 neurons in the trigeminal nucleus. They were found over a depth range of 1200–3100 µm below the dorsal surface of the nucleus (95th percentile range); the average depth was 2200 µm. Most responded to stimulation of the dura and most had receptive fields in the skin (22 in the first trigeminal division, 31 in the second division and one in the third division). The 27 cells positively identified in the histological material were distributed over the nuclear laminae as follows: lamina I, 6; lamina I/IIo, 2; lamina II, 4; lamina III, 5; lamina III/IV, 1; lamina IIo, 2; lamina IV, 4; lamina V, 3.
Fifty-seven neurons in the trigeminal nucleus were recorded while stimulating the skin or dura mater electrically or mechanically. Neurons responded with A-δ latencies to all four types of stimuli. Conditioning stimuli applied to the NRM at 10–500 ms prior to peripheral stimulation suppressed responses to dural electrical and mechanical stimulation and to skin mechanical stimulation, but not to skin electrical stimulation. Suppression was maximal at 30–50 ms and had abated by 500 ms. The degree of suppression was dependent on total charge delivered (current × no. of pulses × pulse width) and was maximal at 5 μC.
Eighty-eight per cent of neurons responsive to dura mater electrical stimulation were suppressed according to the criteria of the critical ratio test, but only 26% of neurons responding to cutaneous electrical stimulation were suppressed. Mean suppression, at an NRM-trigeminal stimulus delay of 50 ms, was 51 ± 2% for dural electrical stimulation and 19 ± 6% for skin electrical stimulation. Sample control and NRM-prestimulated responses are shown in Fig. 2 and mean responses in Table 2.
Effect of NRM stimulation on responses of trigeminovascular sensory neurons to electrical stimulation

Responses of trigeminal neurons to electrical stimulation of the dura mater or of facial skin before and after prestimulation of the nucleus raphe magnus (NRM), delivered 50 ms before stimulation of trigeminal afferents. NRM stimulation suppressed the responses to stimulation of the dura mater much more than it did responses to stimulation of the skin, an effect which was partially reversed by the concomitant iontophoretic application of the 5-HT1B/1D receptor antagonist GR127935 to the neurons.

Sample poststimulus histograms showing the effect of stimulation of the nucleus raphe magnus (NRM) on the responses of trigeminovascular second-order neurons to electrical stimulation of the dura mater (left) or the facial skin (right). Skin or dura mater was stimulated at t = 50 ms (arrows); the NRM stimulus (lower graphs only) was delivered at t = 0 ms. NRM stimulus itself induced discharges in this neuron.
In 17 neurons, the iontophoretic application of the 5-HT1B/1D receptor antagonist GR127935 to trigeminal nucleus neurons, which responded to trigeminal stimulation and in turn were suppressed by NRM prestimulation, reduced the degree of suppression produced by NRM prestimulation from 75 ± 6% to 34 ± 8%, a significant reduction (P < 0.05; Student's t-test, n = 17). Mean responses are shown in Table 2.
Mean responses and suppression by NRM for mechanical stimulation of the skin and dura mater are shown in Table 3.
Effect of NRM stimulation on responses of trigeminovascular sensory neurons to mechanical stimulation

Responses of trigeminal neurons to mechanical stimulation of the dura mater or of facial skin before and after prestimulation of the nucleus raphe magnus (NRM), delivered 50 ms before stimulation of trigeminal afferents. NRM stimulation suppressed the responses to stimulation of the dura mater and the skin. Allowing for the inertia of the stimulating device, these latencies are similar to those seen with electrical stimulation.
D. Effect of CSD on the NRM-stimulus-induced suppression of responses of trigeminal neurons to mechanical stimulation of the dura mater and skin
In seven neurons in five cats, control responses for NRM-induced suppression of
responses of trigeminovascular second-order neurons to mechanical stimulation of
the dura were monitored at 5-min intervals for 20 min. After 20 min, and then at
30-min intervals subsequently, cortical microinjections of 1
In five neurons in three (different) cats, control responses for NRM-induced suppression of responses of trigeminovascular second-order neurons to mechanical stimulation of the skin were monitored as for the dura and four successive waves of CSD induced as for the dura. There was no significant change in the degree of suppression produced by NRM stimulation over the succeeding 2 h.
These results are summarized in Fig. 3.

Effect of four waves of cortical spreading depression (CSD) on the suppression of responses of trigeminovascular second-order neurons to dural mechanical stimulation (♦) and skin mechanical stimulation (▪) by preconditioning nucleus raphe magnus (NRM) stimulation. CSD was initiated at the arrows. seven neurons in five cats for dura mater; five neurons in three (different) cats for skin. CSD gradually antagonized the effects of NRM stimulation on dural responses, but not on skin responses.
Discussion
These experiments have shown that a potential neural network which might account for the initiation of migraine pain exists in animals and behaves in a manner consistent with our hypothesis. We have shown that neuronal activation of the cortex can decrease the neuronal activity of the NRM; that stimulation of the NRM can suppress sensation from the dura mater selectively; and that this suppression involves a serotonergic mechanism.
The apparent origin of the pain of migraine
The pain of migraine appears to arise in the meninges, dura mater and blood vessels of the brain (23, 24). How their sensory innervation comes to be activated in migraine—or if indeed their sensory systems are activated at all—remains a matter for debate. The pain might be due to (i) vasoconstriction directly, perhaps caused by activation of stretch receptors by vascular calibre alteration; (ii) ischaemia and hypoxia resulting from vasoconstriction or vascular shunting; or (iii) release of algogens and inflammatory mediators by some antecedent process such as CSD. A combination of (ii) and (iii) has been suggested recently by Takano et al. (25), although this applies only to migraine with aura. If no such processes actually occur in the sensory innervation, the alternative viewpoint is that the pain signal is a ‘false alarm’ occasioned by some defect in pain perception or modulation. The reason for such a defect is not known, but among the ultimate sources suggested have been the cortex (26) and the brainstem (5), which have both been discussed from the point of view of whether they are a cause or an effect of migraine pain. We suggest, on the basis of the present experiments, that both are causes simultaneously. The current experiments support both the previous cortical and brainstem postulates by showing that migraine triggers can activate cortical neurons and inhibit neurons in the NRM and that this inhibition may lead to selective disinhibition of trigeminovascular sensory transduction involving a 5-HT1 receptor subtype known to be involved in migraine.
The role of the cortex
A range of external and internal influences can produce excessive activation of cortical neurons, and some of these do produce headache. These include epilepsy (27–31), electro-convulsive therapy (32, 33), transcranial magnetic stimulation (34) and transcutaneous low-voltage d.c. stimulation of the cortex (35–37). We have discussed these in more detail elsewhere (1). In these experiments, we chose light flash stimulation and CSD as two methods of activating cortical neurons The former is a trigger for migraine in a subset of patients, and it has long been hypothesized that the latter is responsible for many of the early symptoms in migraine with aura (38, 39). Both are known to cause such activation in the cat model we have used here (18, 40). In these experiments we measured cortical activation indirectly, by monitoring cortical blood flow in the occipital cortex. Blood flow change is a reliable non-invasive indicator of the occurrence of cortical activity, because it is a metabolic consequence of neuronal activity changes and accurately reflects their magnitude and direction. The technique is especially effective for CSD, but we have also used it successfully to monitor the activity of visual cortex neurons following light flash (18). Although CSD is labelled a ‘depression’, it appears to produce most of its prominent neurological symptoms through its initial cortical excitation.
Complex connections exist between the three brain areas that we have examined here. There is evidence to suggest that direct trigeminal afferents may reach as high as the cortex (41), and certainly neurons in the cortex can be activated by a polysynaptic pathway, probably involving the trigeminal nucleus (42–45) and the thalamus (46–48), eventually to reach the cortex (49, 50). In rats, a significantly increased Fos immunoreactivity was found in the upper lip and forelimb regions of the primary somatosensory cortex after noxious stimulation produced by injection of capsaicin in the cisterna magna (51). There is also evidence that cell bodies in brainstem nuclei, especially the dorsal nuclei such as the dorsal raphe but also in the region of the NRM, project to the cortex, including the occipital cortex, and can influence its neuronal activity (52, 53).
The role of the NRM
The cortex in turn projects downward to brainstem nuclei including the raphe nuclei (54) and to the trigeminal nucleus directly (55, 56). Horseradish peroxidase labelling of afferents in the NRM labels large numbers of labelled neurons in the frontal cortex (57). In another study, major targets of the prelimbic area were found to include the PAG region and the raphe nuclei (58). Stimulation in the rostral agranular insular cortex can produce either analgesia or hyperalgesia, and it has been suggested that cerebral cortex activity can change the set-point of pain threshold in a ‘top-down manner’ through such connections (59).
Indirect projections from the cortex to the NRM have also been shown to exist, relaying through the PAG (60), among other nuclei. There is a dense projection of nerve fibres, some of them containing 5-HT, from the PAG to NRM (61–64). It is postulated that the PAG exerts its inhibitory effect on spinal nociceptive functions through the activation of descending serotonergic and noradrenergic pathways that arise from RVM nuclei including the NRM. To investigate the neuroanatomical substrate of this functional link between the PAG and RVM, Odeh et al. (65) labelled axons that project from the ventrolateral PAG to various regions of the pons and medulla oblongata using the anterograde tracing substance, Phaseolus vulgaris leucoagglutinin. They demonstrated that some PAG efferents terminate in the RVM, but a substantial proportion of them project to the intermediate subdivision of the pontobulbar reticular formation. The demonstration of significant PAG projections to NRM provides anatomical evidence for the hypothesis that opiate and stimulation-produced analgesia involves connections from PAG to neurons of NRM which, in turn, inhibit spinal nociceptors (66).
There is some evidence that the brainstem raphe nuclei, including possibly the NRM, receive direct fibre projections from trigeminal and cervical sensory neurons which innervate cranial vessels including the middle cerebral artery (67, 68). There are also indirect projections from the trigeminal nucleus which might relay craniovascular sensory information (69). In the experiments reported here, electrical and mechanical stimulation of the dura mater and skin activated NRM neurons with latencies which were consistent with a monosynaptic pathway. We have not formed an opinion about what role any direct or indirect trigemino-raphe connections might play in nociception in general or headache in particular, but the results do demonstrate that the neural circuitry involved may be complex. The nature of the neurotransmitters utilized by cortico-NRM projection pathways is not known with certainty, but several amino acid transmitters may be involved.
Drugs which decrease the discharge rate of NRM neurons, such as ipsapirone (70), m-chlorophenylpiperazine (MCPP) (71) and 5-methoxydimethyltryptamine (22), precipitate migraine-like headaches with a high degree of reliability (72–74). Conversely, drugs like methiothepin, methysergide and cyproheptadine, which increase the discharge rate of raphe neurons (75), have been used to prevent migraine. All of these drugs are agonists or antagonists at 5-HT receptors, particularly 5-HT autoreceptors on raphe nuclei neurons. Met-enkephalin- and substance P-positive terminals are present on serotonin cells in the raphe nuclei (76), further suggesting involvement with the processing of nociception.
There is ample evidence that NRM neurons, generally containing 5-HT, project to the trigeminal nucleus and the dorsal horn of the spinal cord, and sometimes to both, via axon collaterals (77), and make contact there with incoming sensory fibres and with trigeminothalamic projection neurons (55, 78–84). Gao et al. (85) have suggested that the NRM contains subpopulations of serotonergic cells and that these may serve varied physiological functions. Electrical or chemical stimulation of the NRM influences sensory input at a spinal and trigeminal level and, in particular, selectively inhibits C-fibre input (3). This stimulation inhibits release of sensory neurotransmitters such as glutamate, calcitonin gene-related peptide and substance P (86), actions which can also be produced by serotonin or serotonin agonists (87). A similar inhibition of glutamate release in the cortex is mediated by 5HT1D receptors (88). We believe that the triptans and other 5-HT agonists also act predominantly via reduction of the effectiveness of incoming sensory drive (55), which they may do by reducing the release of sensory neurotransmitters through an action at presynaptic 5HT1D receptors which exist on these fibres (89).
There are three types of neurons in the NRM particularly relevant to the present study: silent ON cells, which respond with an increase in firing prior to withdrawal from the noxious input; spontaneously active OFF cells, which respond by cessation of firing; and Neutral cells in which the discharge rate is unchanged by noxious stimulation (90, 91), but which can sometime be persuaded to switch to ON or OFF neurons depending upon the site of noxious stimulation (e.g. becoming 43% ON, 14% OFF, 43% Neutral after dura mater stimulation.). It is reasonable to assume that the activity of OFF cells produces tonic inhibition of peripheral sensation and that these are the predominant neurons which we have recorded as being inhibited by cortical activation, and they are probably also the neurons which we activated by electrical stimulation. It is also reasonable to assume that ON neurons produce sensory facilitation, and they are known to have an excitatory output to spinal and medullary dorsal horn second-order sensory neurons (not necessarily, but predominantly, those with nociceptor-specific input (92)). In our experiments, a significant proportion of trigeminovascular second-order neurons were initially excited by NRM stimulation, before having their responses to dural stimulation suppressed. This may be because electrical stimulation in the NRM would excite both ON and OFF neurons and hence initially excite, but then inhibit neurons in the trigeminal nucleus. It has also been shown that descending facilitation from RVM nuclei may be responsible for the maintenance of neuropathic pain states (93, 94).
The role of the periaqueductal grey matter
There is evidence that many of the effects attributed to the NRM may be driven from the PAG. In patients undergoing a migraine attack, the area of the brain showing the greatest increase in metabolic activity is in the region of the PAG (95). Migraine-like headache, sensitive to antimigraine drugs, can be triggered by lesions, gliomas, tissue damage, demyelinating plaques or vascular malformations in the PAG (4, 96–98). Neuroanatomical and functional evidence points to a possible direct projection to the PAG from primary dural sensory afferents (99), and electrical stimulation of the dura mater leads to expression of c-fos protein in the PAG of the cat (100). It has also been shown that PAG stimulation inhibits trigeminovascular sensory neurons in the C2 region of the spinal cord (101). The inhibition can be partly blocked at the spinal level by 5HT1 receptor antagonists. Crucial evidence has come from a study by Bartsch et al. (102), in which injection into the PAG of the 5-HT1B/1D agonist antimigraine drug, naratriptan decreased the excitability to electrical stimulation of the dura mater by both Aδ-fibre and C-fibre-mediated responses. Responses to stimulation of the face and cornea were not altered by injection of naratriptan. There is evidence that the PAG exerts its own effects on sensation at least partially via projections to both ON and OFF neurons in the NRM (63, 64).
Work from our laboratories was the first to show that antimigraine drugs with agonist activity at 5-HT receptors might act at the first synapse in the trigeminovascular sensory system (45); our later work has demonstrated that this site of action was specific for processing of sensory information from the dura mater and involved the 5-HT1D receptor subtype (11). Serotonin infusions themselves suppress discharges of first-order sensory neurons in cats (103, 104). In the present experiments we have used the 5-HT1B/1D receptor blocker GR127935 to show that NRM-induced inhibition of trigeminovascular responses is probably mediated by 5-HT1 receptors, consistent with the findings of others and our own earlier results.
Electrical stimulation of the NRM would be expected to excite all neural components in it (ON cells, OFF cells, Neutral cells and fibres of passage). It was therefore surprising to us that CSD was able to interfere selectively with the NRM-induced inhibition of only dural and not cutaneously induced neuronal responses. These results suggest that cortical activation produced by CSD may produce long-term hyperpolarization of a subset of NRM neurons (85), thus both reducing their basal discharge rate and rendering them less susceptible to depolarization imposed by a nearby stimulating electrode and thus facilitating nociception. The change in NRM-induced suppression which we observed with responses to mechanical stimulation of the dura is unlikely to be due to the development of sensitization after hours of recording, for two reasons. First, it is selective (in the same neuron) for dural input. Second, we have shown in unpublished work with TRPV1 receptor mechanisms that sensitization develops in these neurons only after the application of inflammatory mediators and not merely by increased neuronal traffic.
The brainstem nuclei, especially the NRM, would seem therefore to be logical targets for attack by an effective migraine headache preventative drug. If a single neurotransmitter were involved in the descending projections to the NRM, an antagonist to it could insulate the NRM from descending influences, maintain its neuronal discharge rate, and thus keep trigeminovascular sensation under control and patients headache-free.