
Abstract
Short-lasting unilateral neuralgiform headache with conjuntival injection and tearing (SUNCT) syndrome is a rare trigeminal autonomic cephalalgia. We report a patient with prolactinoma and cabergoline-induced SUNCT attacks and the literature is reviewed for a better understanding of the pathophysiology.
Keywords
Introduction
Short-lasting unilateral neuralgiform headache with conjuntival injection and tearing (SUNCT) syndrome is a rare trigeminal autonomic cephalalgia (1). Several cases of secundary SUNCT syndrome, particulary due to posterior fossa abnormalities, have been reported (2). Only a few were associated with pituitary adenomas, especially prolactinomas and growth hormone-secreting tumours (3). The pathophysiology of these headaches is poorly understood, and although a role has been proposed for dural stretch and cavernous sinus invasion, the evidence suggests that this is not the cause and that the headaches are predominantly neurohumorally mediated (4). Dopamine agonists are the therapy for patients with prolactinomas that are not subsidiary for surgery. There are reports of SUNCT attacks induced with these medications (5).
We report a patient with prolactinoma and cabergoline-induced SUNCT attacks and review the literature for a better understanding of the pathophysiology.
Case report
A 22-year-old women had had episodic headache since the age of 15 years compatible with migraine without aura: unilateral location, pulsatile quality, severe intensity, photophobia and lasting 4–6 h. Her grandmother had had a non-functioning adenoma. She presented 6 months before secondary amenorrhoea and galactorrhoea. Her serum prolactin was 283.0 ng/ml (normal 8.7–30.0). All other hormones were at normal concentrations (follicle stimulating hormone 3.5 U/l, luteinizing hormone 1.0 U/l, oestradiol 28 pg/ml, somatomedina C 230.3 ng/ml, adrenocorticotropin hormone 44.20 pg/ml, thyroid-stimulating hormone 1.72 µU/ml, calcium 9.6 mg/dl, insulin 17.2 µU/ml and basal cortisol 35 µg/100 ml), so multiple endocrine neoplasm was ruled out. Magnetic resonance imaging (MRI) scan of the brain revealed a pituitary adenoma on the left side of the gland without extending towards the cavernous sinus (Fig. 1). The patient was given cabergoline 1 mg twice per week. One month later she started with pain strictly confined to the left retro-orbital region and usually lasting <1 min. Physical examination during attacks showed left-sided conjunctival injection, rhinorrhoea and lacrimation without other cranial autonomic features. The pain had a burning quality, started rapidly, was maintained at a plateau phase and then resolved rapidly. There was no nausea, vomiting, photophobia or phonophobia. She felt irritable during the pain and would prefer to move around, but the movement had no effect on the pain. The triggers were stress situations and intense physical exercise without arefractory period. The headaches increased in frequency from three times to more than 10 times per day.

Magnetic resonance imaging scan of the brain reveals a pituitary adenoma on the left side of the gland without extending towards the cavernous sinus.
The patient had previously tried paracetamol 650 mg three times per day, ibuprofen 600 mg three times per day, flunarizine 5 mg daily and indomethacin 50 mg three times per day during a week without any benefit. Six months after starting therapy with cabergoline, her serum prolactin level was normal and the MRI scan of the brain showed that the prolactinoma had greatly decreased in size (Fig. 2).
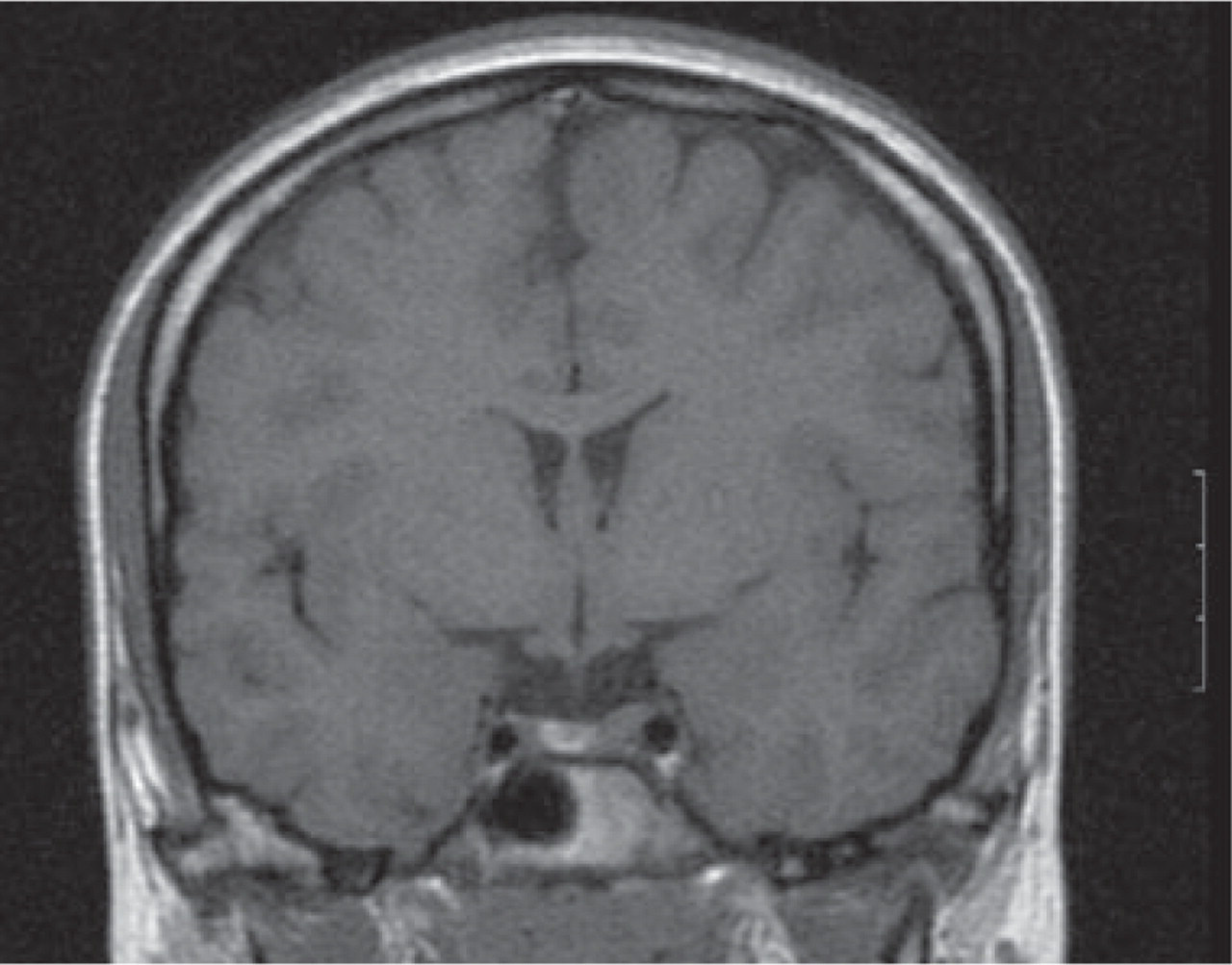
Magnetic resonance imaging scan of the brain shows that the prolactinoma had greatly decreased in size.
Cabergoline was removed and the headache disappeared in 7 weeks, after which she was completely free from headache attacks for 12 months.
Discussion
The phenotype of this headache met the diagnostic criteria for SUNCT (1). Medical treatment of the prolactinoma led to the beginning of headache and the ceasing of treatment with resolution of the pain, which strongly indicates that the prolactinoma was pivotal in triggering the headache, rather than a coincidental pathology. The differential diagnosis between trigeminal neuralgia and SUNCT can be difficult. The pathophysiology of pituitary associated headache is poorly understood, although dural stretch, cavernous sinus invasion and local pressure effects have been proposed as possible mechanisms (6–8). The SUNCT attacks occurred ipsilateral to the side of the tumour, suggesting a role for a mechanical mode of action (4). The cavernous sinus, where trigeminal, parasympathetic and sympathetic nerve fibres are close together, has been discussed pathophysiologically (9). The evidence suggests that this not the cause, because the headache is a recognized feature of small, non-invasive functional tumours, particularly prolactinomas (6, 10), and pituitary tumour size itself is unrelated to headache (11).
Functional MRI in spontaneous attacks has demonstrated activation of the ipsilateral hypothalamic grey (12). It has been suggested that dopamine may play an important role in the pathophysiology of primary headache syndromes (13). In four cases of SUNCT associated with prolactinomas, the administration of dopamine agonists has led to an exacerbation of headache symptoms (5, 14, 15). In a case of SUNCT with prolactinoma, the administration of dopamine agonists was very effective (4). Dopamine is an important hypothalamic hormone that suppresses the secretion of prolactin by the anterior pituitary gland. In prolactinoma, it is the autonomous secretion of prolactin by the pituitary lactotrophs that leads to hyperprolactinaemia. Dopamine agonists share properties with ergot alkaloids (16), which are known to alter the activity of the trigeminovascular system (17). The dramatic exacerbation of SUNCT with dopamine agonists observed in certain cases further suggests that perturbations in the dopamine–prolactin axis may be important in this headache syndrome. The long period between the initiation and cessation of cabergoline and onset and cessation of the headaches could be because the cabergoline did not cause the effect by stimulation of the receptor directly but by neuroendocrine changes in the hypothalamo–pituitary axis that developed in weeks or months, rather than being mediated by the size or invasiveness of the tumour (4). Ipsilateral hypothalamic activation has been demonstrated in primary SUNCT (12) and it is conceivable that specific neuroendocrine pathways involving the dopamine–prolactin axis is capable of activating SUNCT pathophysiology (3). All patients presenting with suspected SUNCT syndrome should be examined for symptoms associated with pituitary neoplasms and by a cranial MRI, including an adequate view of the pituitary, and screening of basal hormone levels.