
Abstract
The aim of this study was to evaluate the function of pain modulating systems subserving diffuse noxious inhibitory controls (DNICs) in primary headaches. DNICs were examined in 24 migraineurs, 17 patients with chronic tension-type headache (CTTH) and 20 healthy subjects by means of nociceptive flexion RIII reflex and the cold pressor test (CPT) as heterotopic noxious conditioning stimulation (HNCS). The subjective pain thresholds (Tp) and the RIII reflex threshold (Tr) were significantly lower in CTTH vs. controls. In controls a significant inhibition of the RIII reflex was observed during the CPT (-30±, P < 0.05). Conversely, migraine and CTTH patients showed facilitation (+31±, P < 0.05 and +40±, P < 0.01, respectively) of the RIII reflex during the HNCS. This study demonstrates a dysfunction in systems subserving DNICs in both migraine and CTTH. Impairment of endogenous supraspinal pain modulation systems may contribute to the development and/or maintenance of central sensitization in primary headaches.
Introduction
A growing body of evidence suggests that abnormal central pain processing may play an important role in the initiation and maintenance of both tension-type headache and migraine (1–6). Several studies have demonstrated an increased sensitivity to pain and low threshold stimuli in chronic tension-type headache (CTTH) (7), suggesting a central sensitization of pain pathways in this condition (3, 5), while clinical, neurophysiological and laboratory studies (2, 8–11) have shown that a temporary hyperexcitability of central nociceptive trigeminal neurons may occur during migraine attacks. It has been postulated that this hyperexcitability is caused by a defective endogenous modulatory antinociceptive system (2, 12). Primary headaches are unique in that they cause severe pain in the absence of an obvious peripheral source of the pain. Furthermore, migraine pain is associated with an enhancement of sensory function and modulated by emotional and physiological mechanisms (13). These phenomena suggest a pathogenic contribution of CNS pain-modulating pathways, a hypothesis supported by functional imaging studies (14–16).
A selective, task-specific activity of the supraspinal pain modulation system may be investigated dynamically by examining ‘diffuse noxious inhibitory controls’ (DNICs) (17). DNICs may be defined as the inhibition of nociceptive neurons in the spinal and trigeminal dorsal horns produced by a noxious stimulus applied to any body region remote from the neurons’ excitatory receptive field (18). The anatomical and neurophysiological bases of DNICs have been studied in detail in both animals and humans (19, 20) and their effects can be investigated by evaluating their inhibitory action on subjective psychophysical measurements and/or nociceptive reflexes (19).
The nociceptive flexion reflex (NFR) is a reliable and objective tool for exploring pain control systems in humans (20). The threshold and amplitude of the RIII reflex are closely related to the threshold and amplitude of the concomitant pain evoked by the electrical stimulus and the RIII reflex has been reported to be significantly inhibited by activation of DNICs (17, 19–21). The aim of this study was to evaluate the function of pain modulating systems subserving DNICs in primary headache (migraine and CTTH) by studying the effects of heterotopic noxious conditioning stimulation (HNCS), in the form of the cold pressor test (CPT), on the NFR.
Methods
Subjects
Forty-one patients, diagnosed with migraine without aura (MoA) (n = 24; mean age 36 ± 12 years, 12 males and 12 females) or with CTTH (n = 17; mean age 32 ± 12 years, eight males and nine females) according to the 1988 International Headache Society (IHS) classification criteria (22), participated in the study. The IHS Classification Committee's revised criteria (23) do not alter these diagnoses. Twenty healthy volunteers (age 32 ± 7 years, six males and 14 females) served as controls.
The CTTH patients had no concomitant migraine attacks or migrainous exacerbations. They had no personal or family history of migraine and none of them had ever been medication overusers.
The mean attack frequency was 3.2 ± 1.1 per month in the MoA patients and 28.4 ± 2.3 headache days/month in the CTTH patients. Mean disease duration was 13 ± 7.4 years in the MoA group and 8.9 ± 4.8 years in the CTTH patients.
The headache patients had not been receiving prophylactic therapy during the 2 months leading up to the study and were required to have been drug-free for at least 24 h. Eleven CTTH patients were examined on headache-free days. The remaining six CTTH patients, affected by daily headaches, were examined during a painful period and rated their headache severity as mild on a 4-point verbal scale. All the migraine patients were examined during a pain-free period at least 48 h after their last migraine attack. On the day following the examination, the patients were contacted by telephone and excluded from the study if found to have experienced a migraine attack within 24 h of the recording. Since there exist neurophysiological findings that support the hypothesis of varying pain control system modulation across the menstrual cycle (24), female patients and controls were matched for cycle phases. All the subjects were made fully aware of the experimental procedure and gave their informed consent before entering the study. The study was approved by the local ethics committee.
Eight healthy volunteers underwent the examination twice, at intervals of no less than 2 days, in order to evaluate the reproducibility of the RIII response following the CPT.
The subjects were eligible to be included in the study if they met the following criteria: (i) no history of any medical or psychiatric illness; (ii) no clinically significant history of neurological disease; (iii) no abnormalities emerging on neurological examination.
No subject with a Hamilton Rating Scale for Anxiety and/or Depression (HRSA and/or HRSD) score of over 20 was included.
Experimental procedure
The NFR was investigated according to a previously described method (19, 21, 24). Briefly, the sural nerve was stimulated with a pair of surface electrodes placed on degreased skin at the retromalleolar site. The stimulus consisted of five rectangular pulses (each of 1 ms duration) lasting 20 ms at a rate of 300 Hz delivered by a constant current stimulator at random intervals (5–15 s). Muscular response (RIII reflex) was recorded electromyographically from the ipsilateral biceps femoris muscle (capitis brevis) via a pair of surface electrodes. The minimal intensity of current needed to elicit a reflex response and to maintain it for 80–90% of the stimulus was taken as the reflex threshold (Tr). The stimulus intensity was adjusted to 20% above the Tr and was kept constant throughout the experiment. The electromyographic recordings were fed to an oscilloscope and to a computerized system. In accordance with the literature, the time windows used to differentiate the RII from the RIII components were 30–70 ms and 80–130 ms, respectively (19–21). Each RIII response was full-wave rectified and integrated between set points from 80 to 130 ms. Ten reflex responses were recorded and the averaged RIII reflex areas were calculated by means of a computerized method. The time window was adjusted in each case to avoid integration of any component of the tactile reflex (RII).
In order to minimize noise signal interference, visual inspection was used to verify all the assessments and EMG traces with a peak amplitude above 20 µV in the prereflex interval were excluded.
The subjective pain sensation elicited by the sural nerve stimulation was graded on a 10-level visual analogue scale (VAS; 0 = no pain to 10 = intolerable pain). The subjects were asked to report the subjective threshold for pain perception (Tp), which was defined as level 3, while levels 1 and 2 represented tactile sensations. The right side was examined in all patients.
In order to explore the pain modulating systems subserving DNICs, we investigated the effects of HNCS, in the form of the CPT, on the NFR. When prompted by the experimenter, the subjects immersed their contralateral hand, for 5 min, in a circulating water bath kept at 5–6°C. Ten reflex responses were recorded before the CPT (baseline recording), during the first 2 min (CPT1) and during the second part (CPT2, 3rd to 5th minute) of the CPT and during the 2 min immediately after the conditioning stimulation. In 21 subjects (seven migraineurs, seven CTTH patients and seven controls) the neurophysiological measurements were recorded in an additional neutral condition (dipping the hand in water at 27°C). To avoid any possible sensitization of the skin receptors, the hand was dried after removal from the water and a resting period of more than 20 min was allowed between each sequence.
Since activation of DNICs induces a significant, reproducible, concomitant modulation of NFR area and pain sensation without affecting the NFR latency and duration (17, 19–21), data regarding the latter parameters were not analysed. Differences in the NFR area and subjective pain sensation during and after the CPT or neutral session were presented as percentages of baseline values.
Neurophysiological examinations were always performed by the same operator, who was blind to the subjects’ diagnoses.
Statistical analysis
All analyses were performed on a personal computer using SPSS for Windows, version 11.0 (SPSS Inc., Chicago, IL, USA). The data were expressed as mean values ± SD.
Results
No significant correlation was found between clinical parameters (age, sex, age at onset, duration of disease, attack frequency) and neurophysiological data. Moreover, no correlation was observed between HRSA and HRSD scores and neurophysiological parameters.
No sex-related differences were observed between the patient and control groups. The NFR latency and mean NFR area of the headache patients did not differ significantly from those of the controls (Table 1). The subjective pain thresholds (Tp) and the NFR thresholds (Tr) were significantly lower in CTTH patients than in controls (Table 1).
Nociceptive flexion reflex parameters in headache patients and controls (mean ± SD)

Tp, Pain threshold; Tr, reflex threshold; CTTH, chronic tension-type headache.
P < 0.01 vs. controls (one-way
Figure 1 shows the effects of the CPT on the NFR. In the controls, a significant inhibition of the NFR was observed during the cold stimulation (CPT1, first 2 min, −35%, P < 0.01; CPT2, 3rd to 5th minute, −30%, P < 0.05). In contrast, the headache patients demonstrated a lack of inhibition. In fact, migraine and CTTH patients showed no inhibition, but facilitation of the RIII reflex during the HNCS (Fig. 2), a phenomenon that was more evident in the CTTH group (CTTH, CPT1 +40%, P < 0.01; CPT2 +31%, P < 0.05; MoA, CPT1, +31%, P < 0.05; CPT2, +14%, P > 0.05). It should be noted that the controls demonstrated maximum inhibition during the first 2 min of the CPT, after which, during the remaining minutes of the CPT and after the test, inhibition decreased. A different pattern (maximum facilitation during the CPT, gradually decreasing in the post-conditioning period) was found in both headache groups.
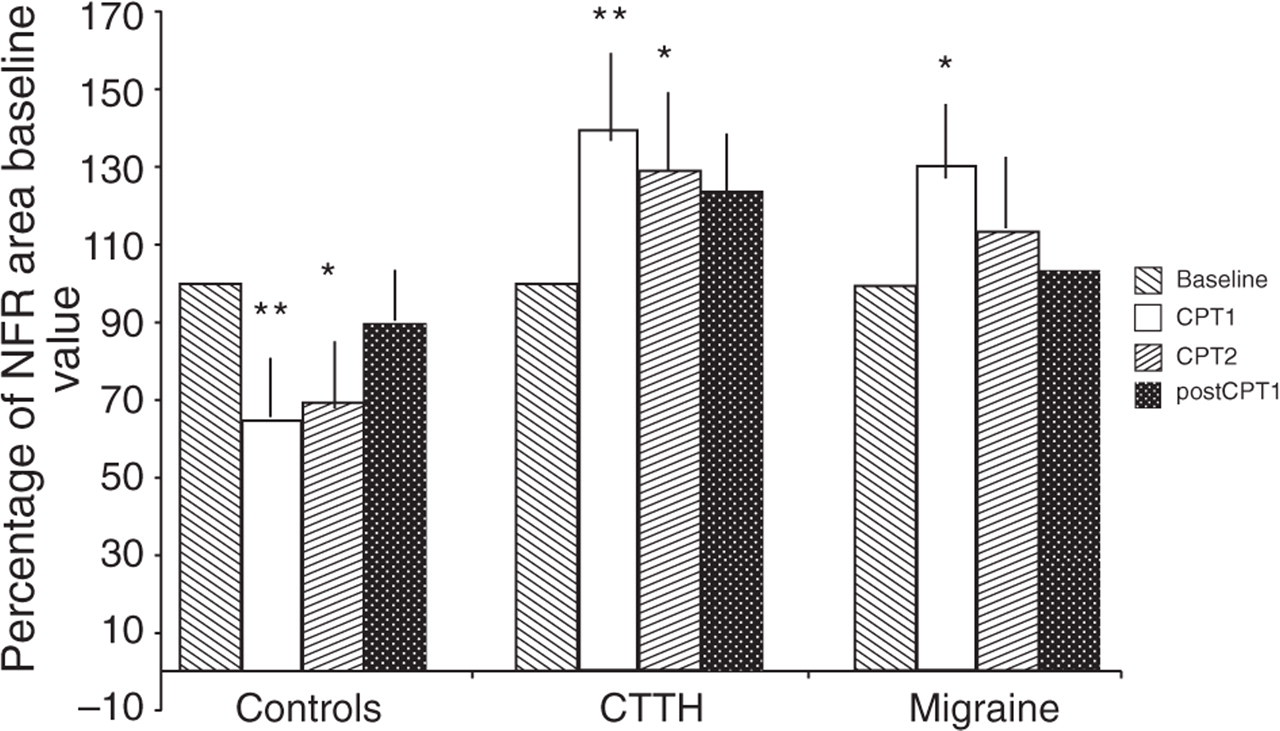
Effects of the cold pressor test (CPT) on mean reflex area in controls and patients. The paired t-test was used to make comparisons with the baseline values expressed as 100% (∗P < 0.05; ∗∗P < 0.01). Data are expressed as percentages of baseline values recorded during the 2-min control period and each histogram represents the mean ± SD. In controls the RIII reflex area was significantly decreased during the first 2 min (CPT1, P < 0.01) and second part (CPT2 3rd to 5th minute, P < 0.05) of the CPT. In chronic tension-type headache (CTTH) and migraine patients the mean reflex area was significantly increased during CPT1 (P < 0.01 and P < 0.05, respectively) and the facilitatory effect persisted during the CPT2 in the CTTH group (P < 0.05). After CPT, a gradual return to baseline values was observed in both groups. NFR, Nociceptive flexion reflex.

RIII reflex area before (upper trace) and during (lower trace) the first 2 min of the cold pressor test (CPT1) in a representative chronic tension-type headache subject (50 ms, 100 µV per division). The CPT induced an increase, instead of a decrease, in the nociceptive flexion reflex (arrows) area (EMG average of 10 reflex responses).
The results of subjective pain perception during the CPT paralleled those of the RIII reflex area in both the control subjects and the headache patients (Table 2).
Summary of changes in subjective pain perception in response to sural nerve stimulation (Visual Analogue Scale 0–10) after activation of diffuse noxious inhibitory controls (mean ± SD)

CPT1, Cold pressor test (first 2 min); CPT2, cold pressor test (3rd to 5th minute).
Paired t-test P < 0.05 vs. baseline.
In the non-noxious session no group showed significant differences vs. controls in either RIII reflex area or subjective pain perception.
The Cronbach α measure was 0.71, indicating an acceptable level of test–retest reliability of CPT effects on the RIII response.
Discussion
In this study we evaluated, in migraine and CTTH patients, the function of circuits subserving ‘diffuse noxious inhibitory controls’ by investigating the effect of HNCS on the NFR of the lower limbs.
The use of a spinal model of nociception is justified by the consideration that the descending pain modulatory influences originating from the brainstem (see below) in response to noxious stimuli applied to any part of the body distant from the neurons’ excitatory fields are common to trigeminal and spinal levels and allow the DNIC system to exert a powerful, widespread modulatory influence on the activity of all trigeminal and spinal convergent wide dynamic range neurons participating in nociception (17, 18, 25).
In addition, even though a DNIC effect has been demonstrated for nociceptive trigeminal-mediated responses (i.e. blink reflex, trigemino-cervical reflex) (26–28), the NFR is better standardized and more sensitive to changes in gain of nociceptive transmission, such as those induced by HNCS, than neurophysiological reflexes used to investigate nociception in the craniofacial region, because at this level, pain-induced motor response recruitment is more variable and needs to be integrated with more complex motor activities resulting from visual, acoustic, vestibular and other stimuli (26, 27).
As expected, in normal subjects the HNCS depressed both the NFR and the associated painful sensation (21, 29). By contrast, in both MoA and CTTH patients, the CPT did not produce any DNIC-like effect but, instead, a trend towards a facilitation of the NFR. These findings suggest a dysfunction in systems subserving DNICs in headache patients.
It is well known that DNIC circuits may also have a facilitatory influence on dorsal horn nociresponsive neurons (25, 30) and from this perspective the findings in our headache patients may be interpreted as the result of the prevalence of facilitatory influences activated in the setting of an acute noxious stimulation, in this case the CPT.
Possible explanations for such impaired DNIC function can be inferred from the known anatomo-functional organization of DNIC systems and from data provided by other studies of pain modulatory systems in migraine and CTTH.
Anatomical and electrophysiological studies, performed in both animals and humans, indicate that DNICs originate from a complex spino-bulbo-spinal loop, specifically activated by A-delta and C peripheral fibres (17, 18). The ascending and descending limbs of this loop travel, respectively, through the ventro-lateral (crossed) and dorso-lateral (uncrossed) funiculi (17, 18, 29, 31). The brainstem, namely the medullary reticular formation, is Key neuronal link of the loop subserving DNICs (18, 31). The inhibitory bulbospinal pathways are serotoninergic (17, 32) and inhibit all the activities of the convergent wide dynamic range neurons at trigeminal and spinal levels, but additional non-serotoninergic mechanisms are also involved (17, 32).
Since in our patients there was no evidence of an impairment of the afferent and efferent pathways subserving DNICs, the most likely explanation of our findings is defective functional activity of supraspinal structures involved in DNIC activities. Interestingly, inhibitory effects of DNICs on both motor response and subjective sensory pain perception were impaired in the MoA and CTTH patients.
Abnormal central processing of sensory information in CTTH has been documented in several studies (3, 7, 33). CTTH patients have been found to be hypersensitive to several types of stimulus applied at both cephalic and extracephalic non-symptomatic locations, indicating that general pain sensitivity is affected at a supraspinal level (3, 7). This may explain the reduction in subjective pain and NFR threshold observed in our study (33). A recent evaluation of the stimulus–response function for pressure vs. pain has shown that CTTH may be caused by qualitative changes in the central processing of sensory information, probably a central sensitization of spinal dorsal/trigeminal nuclei (34). In this regard, CTTH is similar to other musculoskeletal disorders (35). A dysfunction of DNIC-like mechanisms has been found, using different experimental paradigms, in patients with temporo-mandibular disorders (36), chronic low back pain (37), fibromyalgia (38) and painful osteoarthritis (39). These findings have led different authors to conclude that these patients were unable to activate endogenous supraspinal pain modulation (39). Our findings suggest that CTTH shares with other forms of chronic pain an impairment of CNS regulatory systems, possibly secondary to a prolonged nociceptive input from the periphery. It remains to be established whether DNIC dysfunction is an aspecific neurophysiological effect of chronic pain, or plays a pathogenic role in the development of central sensitization of nociceptive pathways.
In this study, we did not investigate a chronic pain control group, thus aspecificity of the findings cannot be ruled out. It is noteworthy that the migraine patients showed comparable DNIC dysfunction, in the absence of increased pain sensitivity, when studied in a pain-free period. Several laboratory and human studies have shown that during a migraine attack a temporary hyperexcitability of central nociceptive trigeminal neurons may occur (8–10). Burstein has hypothesized that this sensitization may develop as a result of a dysfunction (prevalence of facilitatory vs. inhibitory activities) of descending pathways implicated in the control of the trigeminovascular nociceptive system (2). Neurophysiological evidence for central sensitization in migraine patients also in the interictal phase has recently been reported (11).
In a recent study, Welch et al. (16) demonstrated selective and progressive impairment of iron homeostasis in the periaqueductal grey (PAG) of both migraine and chronic daily headache patients. Physiological and anatomical studies have shown that the PAG exerts a complex modulatory influence on DNICs that is mainly, but not exclusively, mediated by opioid receptors, most probably encephalinergic ones (17, 40). Thus, a permanently dysfunctional PAG may be implicated in the generation of migraine (13, 14, 16) and the inhibition of cooperating modulatory systems subserving the DNICs. As for CTTH, it remains to be established whether DNIC hypoactivity has a role in inducing or maintaining migraine attacks.
From what has been said thus far, it would seem likely that abnormal supraspinal modulation and central sensitization are common denominators of CTTH and migraine (6), although their basic pathophysiological mechanisms are different. In the former, the increased facilitation and decreased inhibition of pain transmission at brainstem level may be secondary to prolonged nociceptive inputs from peripheral myofascial tissues; in the latter, the same pattern of supraspinal pain modulation may be the result of a primary dysfunction of brainstem nuclei.
Our findings are in line with those of Ashina et al. (41), who have demonstrated that systemic administration of the nitric oxide (NO) donor glyceryl-trinitrate in patients with CTTH, as in migraineurs, induced a biphasic nociceptive response with an immediate and delayed headache, the latter reflecting the phenotype of the primary headache. These authors concluded that a pre-existing facilitation of distinct nociceptive central pathways could be enhanced by NO-induced central sensitization (41).
Basic pain research has shown that endogenous NO production plays an important role in the development of central sensitization of nociceptive pathways (42, 43). NO acts as neurotransmitter or neuromodulator at different levels of the central pain pathways, but in humans its role in nociceptive modulation is largely unknown (12). In a recent paper Srikiatkhachorn et al. (44) demonstrated, in an animal model, that the effects of 5HT2A receptor activation on the trigeminovascular NO pathway resemble those observed after glyceryltrinitrate infusion. These authors suggest that activation of the 5HT2A receptor can facilitate the nociceptive process by inducing NO formation and sensitivity to pain. As mentioned before, DNIC efferents are mediated by serotoninergic pathways (17, 32). Impairment of 5HT modulatory pathways has been widely demonstrated in both migraine (45) and CTTH patients (3) and represents a possible biochemical mechanism underlying the observed findings. However, this must be regarded only as a preliminary hypothesis, with further studies being needed to establish the exact pathogenic role of serotoninergic dysfunction in modulating the pain of several types of primary headache.
It has recently been demonstrated that DNIC effects on psychophysical measures of second pain are gender specific (46). The lack of a pain-inhibiting mechanism in women, possibly related to a defective distraction effect of conditioning stimuli, may represent a predisposing factor in the development and maintenance of chronic painful conditions such as fibromyalgia (46). In our present study, we, like other authors who used the NFR (relying on A-delta input) as an outcome measure (47), failed to find any gender-related difference in DNIC effects, which suggests that gender differences in pain modulation are limited to the perception of painful sensations specifically evoked by C-fibres.
It could be argued that the DNIC dysfunction observed in headache patients may be the result of a non-specific effect such as differential changes in attention or arousal (46). In a recent paper, Reinert et al. (48), demonstrated that an HNCS activating the DNICs did not significantly affect the neurophysiological correlates of vigilance and attention and that the analgesic effect was based mainly on activation of specific inhibitory pain control systems. It is thus probable that these mechanisms make no major contribution.
It has been well established that, between attacks, migraine sufferers are characterized by a defective habituation (or even potentiation) of cortical and subcortical responses during stimulus repetition (49). Deficient habituation has been found for different sensory modalities, including nociceptive stimuli, and it could be responsible for area/amplitude differences observed between migraineurs and controls obtained when averaging multiple responses. In our study, the temporal characteristics of the stimulation (low frequency, random intervals) did not induce any short-term habituation effect (21). The contribution of a possible long-term defective habituation effect in migraine patients may be excluded on the basis of the consideration that the increase in the RIII reflex area did not persist after CPT, suggesting that the observed area differences were specifically related to the activation of the DNICs.
In summary, we have demonstrated that pain modulatory systems subserving DNICs are impaired in both migraine and CTTH. Additional research is necessary to further our understanding of the pathophysiological role of supraspinal modulatory networks in primary headaches and their contribution to the development or maintenance of central sensitization.