
Abstract
The molecular basis of migraine is still not completely understood. An impairment of mitochondrial oxidative metabolism might play a role in the pathophysiology of this disease, by influencing neuronal information processing. Biochemical assays of platelets and muscle biopsies performed in migraine sufferers have shown a decreased activity of the respiratory chain enzymes. Studies with phosphorus magnetic resonance spectroscopy (31P-MRS) have demonstrated an impairment of the brain oxidative energy metabolism both during and between migraine attacks. However, molecular genetic studies have not detected specific mitochondrial DNA (mtDNA) mutations in patients with migraine, although other studies suggest that particular genetic markers (i.e. neutral polymorphisms or secondary mtDNA mutations) might be present in some migraine sufferers. Further studies are still needed to clarify if migraine is associated with unidentified mutations on the mtDNA or on nuclear genes that code mitochondrial proteins. In this paper, we review morphological, biochemical, imaging and genetic studies which bear on the hypothesis that migraine may be related to mitochondrial dysfunction at least in some individuals.
Keywords
Introduction
Mitochondria are bacterium-sized organelles found in all mammalian cells (1, 2). They play a central role in a variety of cellular functions including energy generation, reactive oxygen species (ROS) production, regulation of apoptosis and control of calcium homeostasis (3).
Mitochondrial diseases are a heterogeneous group of disorders attributed to mitochondrial dysfunction (4–6). Over the past two decades, technological advances in the field of molecular genetics have been applied successfully to the study of these diseases. A wide variety of neurodegenerative disorders have been associated to mutations of the mitochondrial DNA (mtDNA) as well as to mutations of nuclear DNA (nDNA) genes encoding mitochondrial respiratory chain subunits or other mitochondrial proteins (3–6).
Although advances have been made in the study of the familial aggregation of migraine, and in the search for the migraine genes (7–12), the molecular basis of migraine is far from being completely understood. Several lines of evidence suggest that at least some subtypes of migraine may be related to a mitochondrial defect. In this paper we review morphological, biochemical, imaging and genetic studies that investigated the mitochondrial dysfunctions in migraine sufferers. We critically discuss the limitations of positive studies, as well as the flaws of some negative studies. We close by correlating these findings with our current knowledge of migraine pathophysiology.
Morphological studies
It is well known that structural mitochondrial abnormalities in disorders such as the mitochondrial encephalomyopathies affect mainly non-actively dividing tissues, such as the skeletal muscle and neurons (13–15). Ethical reasons have so far limited histological and/or ultrastructural examination of both tissues in migraine sufferers. However, ragged-red fibres (RRFs) and cytochrome-c-oxidase (COX) negative fibres have been found in skeletal muscle of some patients with migraine with prolonged aura (MPA) (16, 17) and familial hemiplegic migraine (FHM) (18) (Table 1). RRFs are muscle fibres with an abnormal number of subsarcolemmal mitochondria, and are still considered ‘the most informative light microscopic alteration’ (13) of mitochondrial diseases. Although an increased amount of fat occurs in certain COX deficiencies (19, 20), COX-negative fibres are considered ‘the histochemical signature’ of most mitochondrial encephalomyopathies (13, 15, 21, 22).
Morphological studies in migraine
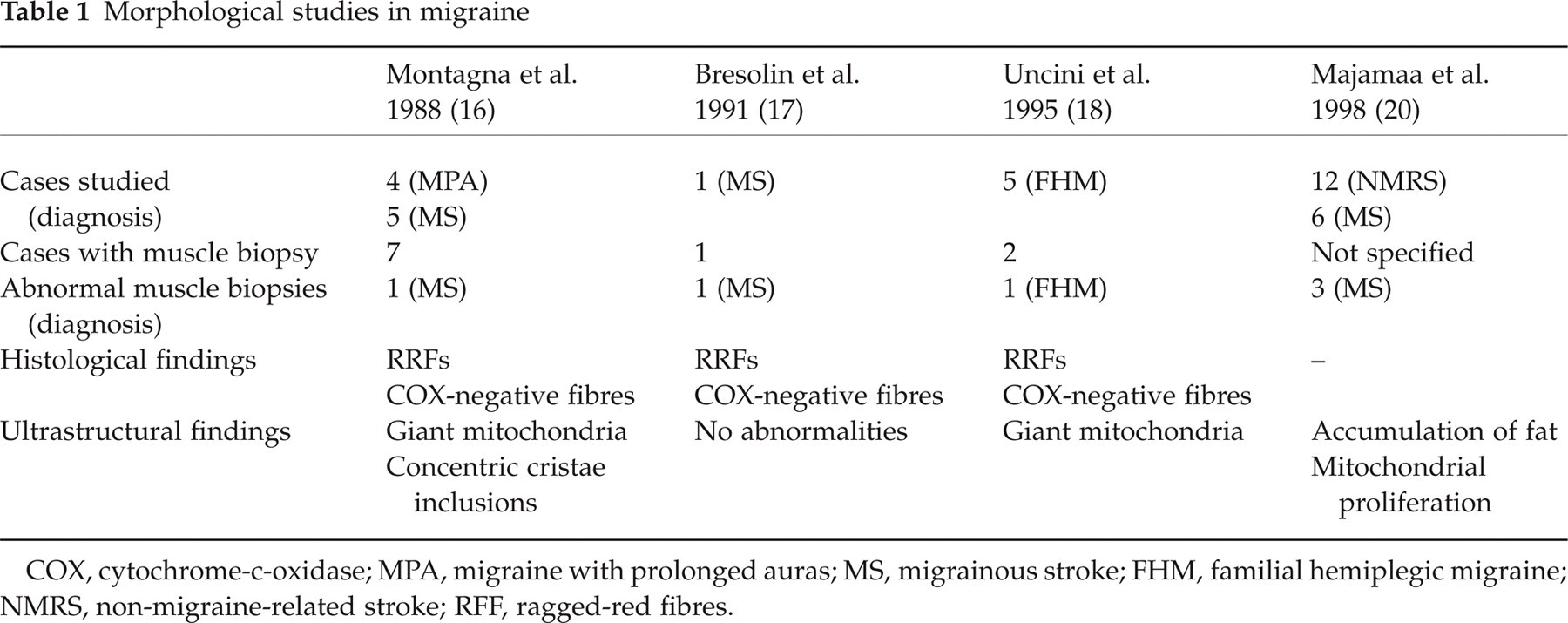
COX, cytochrome-c-oxidase; MPA, migraine with prolonged auras; MS, migrainous stroke; FHM, familial hemiplegic migraine; NMRS, non-migraine-related stroke; RFF, ragged-red fibres.
Electron microscopical studies have shown clusters of giant mitochondria with paracrystalline inclusions in migraine sufferers (16, 18). Some ultrastructural changes, such as an increased amount of fat (probably reflecting a general impairment of intermediate metabolism, as in certain forms of COX deficiency) (23–25), and accumulation of subsarcolemmal or interfibrillar mitochondria, have been found also in muscle fibres of patients with migrainous stroke (MS) (24). However, no evidence of the most common mtDNA mutations at genetic molecular analysis was found in these cases (16, 18, 24). It must be noted that even if ultrastructural alterations of mitochondria are not restricted to syndromes resulting from primary errors of mitochondrial metabolism, they still provide clues to the diagnosis of mitochondrial diseases (23). These changes, concerning the number (excessive proliferation of mitochondria) or the structure (alteration of number and orientation of cristae; intramitochondrial paracrystalline inclusions, etc.) of these organelles represent, in fact, the manner that mitochondria utilize to respond to a metabolic perturbation (23, 25, 26). Interestingly, they are more frequent in disorders of the respiratory chain or of oxidation-phosphorylation coupling (25).
Biochemical studies
Biochemical evidence supporting a deficit in the muscular and/or cerebral mitochondrial energy metabolism has been provided by studies analysing the concentration of lactic acid in the blood and cerebrospinal fluid (CSF) of migraine sufferers. Skinh⊘j, in 1973, was the first to demonstrate that lactate levels are increased in the CSF during migraine attacks (27). Okada et al. examined the lactic and pyruvic acid levels in the plasma of subjects affected by migraine with aura (MA) (
Increased concentration of lactate in the blood (at rest or after a moderate exercise) and in the CSF is still considered an important, though non-specific, laboratory hallmark of mitochondrial disorders, indicating a defective oxidative metabolism (25). Lactic acidosis, in fact, is due to an impaired utilization of pyruvate in Krebs cycle. This defect causes consequently an increased concentration of pyruvate that is reduced to lactate by lactate dehydrogenase (25). An increased glycogen utilization and glycolytic activity further contributes to the rise of lactate in tissues with defective aerobic metabolism (25). Hence, the finding of lactic acidosis in migraine sufferers first points to a generalized metabolic dysfunction that, according to the degree of impairment of the oxidative metabolism, may became evident during or between attacks.
Littlewood et al. provided evidence for a generalized metabolic dysfunction in migraine, by demonstrating a reduced activity of some mitochondrial enzymes, such as monoamine-oxidase (MAO) and succinate-dehydrogenase, in the platelets of 48 patients with MoA and MA (29). Montagna et al. reported that, during the exercise, the blood levels of lactate rose significantly in four out of four investigated patients with migraine with prolonged aura and in five out of five patients with MS (16). On muscle biochemical analyses, the same authors reported depression of some mitochondrial enzymes such as COX (mean values = 41.42 nmol substrate used per min/mg tissue; normal values = 59.45 ± 10.12; mean reduction = 30%), NADH-cytochrome-c-reductase (mean values = 35.19 nmol substrate used per min/mg tissue; normal values = 56.24 ± 10.12; mean reduction = 37%) and succinate-cytochrome-c-reductase (mean values = 12.60 nmol substrate used per min/mg tissue; normal values = 17.35 ± 3.09; mean reduction = 27%) in seven out of nine investigated migraine sufferers (16). Interestingly, such biochemical changes were restricted to enzymes of the respiratory chain, since citrate-synthetase, an enzyme of the Krebs cycle present in the mitochondrial matrix and not coded by the mtDNA, remained normal (16). The authors considered these findings indicative of an altered oxidative metabolism in the skeletal muscle (16).
A more generic impairment of the mitochondrial metabolism, rather than specific respiratory chain defects, was shown by Sangiorgi et al. in the platelets of 40 patients with MA and 40 with MoA, collected between the attacks (30). Besides a deficit of NADH-dehydrogenase and COX, these authors found a statistically significant decreased activity of citrate-synthetase in both patient groups (30).
A recent study presented the cases of two female children suffering from MoA, recurrent fatigue, muscle cramps and multiple side-effects from their migraine prophylactic treatment (31). Both had low levels of carnitine, a major component of the mitochondrial fatty acid transportation system (31). Muscle biopsy in one patient demonstrated a partial carnitine palmityltransferase II deficiency (31). In a prior study, headache and carnitine deficiency had been reported in a subject on haemodialysis for renal failure (32). Interestingly, in these three subjects headache response was favourable after carnitine replacement (31, 32).
Phosphorus magnetic resonance spectroscopy studies
Phosphorus magnetic resonance spectroscopy (31P-MRS) is a non-invasive method that analyses the efficiency of ATP production and allows
MRS studies showing impairment of the brain energy metabolism in migraineurs have brought additional evidence about the relationship between migraine and mitochondrial dysfunction. Watanabe et al. reported elevated lactic acid levels during the interictal period, in the occipital visual cortex of five migraineurs (two with MA, one with basilar migraine, one with MS and one with migraine with prolonged aura and MS) out of six investigated (38). Welch et al. measured the brain energy phosphate metabolism during migraine attacks in 12 patients with MoA and eight patients with MA (39). They found significant reduction of PCr and increased concentration of Pi resulting in a low PCr/Pi ratio, indicating a low availability of free cellular energy in MA (but not in MoA) sufferers (39).
Better to assess specific vascular and humoral modifications that happen during migraine attacks, subsequent studies using single voxel 31P-MRS have been performed during attack-free periods. The following interictal brain abnormalities in occipital lobes are common to different forms of migraine, such as MoA (39, 40), MA (38, 41), FHM (18), migraine with prolonged aura or MS (17, 42, 43, see Table 2 for details):
Most common 31P-MRS brain abnormalities found in migraine sufferers between attacks and in patients with mitochondrial encephalomyopathies

MoA, migraine without aura; MA, migraine with aura; FHM, familial hemiplegic migraine; MPA, migraine with prolonged aura; MS, migrainous stroke; CPEO, chronic progressive external ophthalmoplegia; MELAS, mitochondrial encephalopathy, lactic acidosis and stroke-like episode; LD, leigh's disease; MERRF, myoclonic epilepsy with ragged-red fibres; LHON, Leber's hereditary optic neuropathy; MM, mitochondrial myopathy; RP, retinitis pigmentosa; PCr, phosphocreatine; Pi, inorganic phosphate; ADP, adenosine diphosphate;
2 CPEO; 1 MELAS; 1 LD; 1 MERRF.
12 CPEO; 6 LHON; 5 MM; 1 MS; 1 MERRF; 1 MELAS; 2 RP.
Low concentrations of PCr and increased Pi content resulting in a depressed PCr/Pi ratio (low availability of free energy in the cell) (40, 43).
Increased free ADP concentration, indicating that the brain tissue is working at a higher metabolic rate and has a lower energy reserve (43).
Increased
Altogether, these abnormalities are compatible with an unstable metabolic state of the brain and a decreased ability to cope with further energy demand (36, 41). It is noteworthy that the same findings have been found in patients with some mitochondrial encephalomyopathies (35, 44) (Table 2), suggesting that they may represent a cerebral 31P-MRS pattern that could be typical of mitochondrial disorders.
Lodi et al. found further evidence of interictal mitochondrial dysfunction in the occipital lobe of patients with different migraine subtypes (seven MS; 13 migraine with prolonged aura; 37 MA or basilar migraine; 21 MoA) (45). These authors showed a decreased amount of energy released by the reaction of ATP hydrolysis in all subgroups of migraine (45). Interestingly, the severity of these findings correlated with the intensity of the clinical phenotype: subjects with MS displayed the lowest mean value of free energy released by the reaction of ATP hydrolysis (−80.3 ± 0.20 KJ/mol; controls =−81.8 ± 0.08 KJ/mol), while those with MoA showed the highest value (−80.9 ± 0.13 KJ/mol; controls =−81.8 ± 0.08 KJ/mol) (45).
More recently, Boska et al. studied multiple brain cortex regions in subjects with MoA (
It is important to point out that the abnormalities found in the above discussed studies may be caused by a primary mitochondria dysfunction, or just reflect the state of brain hyperxcitability that happens in migraine. In a critical review, Montagna et al. summarize that the brain energy metabolism is abnormal in all major subtypes of migraine, both during and between attacks (36), but ‘whether impaired energy metabolism is primary or secondary to another causative factor (reduced Mg, an ion channel disorder, etc.) remains unclear’ (47).
Genetic studies
The familial aggregation of migraine was first reported by Liveing, in 1873 (7–12, 48). Recent studies have demonstrated that up to 75% of the FHM cases have missense mutations in the chromosome 19p13 gene, CACNA1A, encoding the α subunit of the voltage-gated P/Q-type calcium channel (49, 50). More recently, four distinct pedigrees (two Italian, one Dutch and one Dutch-Canadian) with a rare form of FHM (FHM type 2) have been associated with missense mutations in a chromosome 1q23 gene, ATP1A2, encoding the α2 subunit of the Na+,K+-ATPase, an integral plasma membrane enzyme that utilizes ATP hydrolysis to extrude three intracellular Na+ ions in exchange for two extracellular K+ ions (51–53). Na+ gradient, provided by the Na+,K+-ATPase pump, is essential to several cellular functions, such as control of cellular volume, generation of action potentials and termination of synaptic activity through astrocytic removal of glutamate from the synaptic cleft (52, 54). All these findings suggest that besides the genetic defects, also a failure of the ATP synthesis, perhaps due to mitochondrial dysfunction may impair the activity of the Na+,K+-ATPase pump, increasing the cortical excitability. Loss of Na+,K+-ATPase function, in fact, may render the neurons hyperexcitable by several mechanisms such as rise in extracellular K+, increase of intracellular Na+ and decreased astrocytic removal of glutamate from the synaptic cleft (52).
Further evidence that an alteration of the mitochondrial genetic background might be involved in migraine aetiology includes:
Some clinical features of recurrent attacks of prolonged migrainous headache overlap with those of MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes), an inherited mitochondrial encephalomyopathy associated, in 80–86% of the cases, with an A→G transition at position 3243 of mtDNA (55–58). ‘Migraine-like headache’ represents the predominant symptom at onset in 50% of patients with MELAS and has been noted in 73–92.5% of cases during the entire course of the disease (58–61). Migraine-like attacks in MELAS are generally severe and prolonged, and sometimes occur in clusters lasting a few days which often precede strokes (61). Interestingly, headache has sometimes been reported as the only clinical feature in oligosymptomatic maternal relatives of patients with MELAS (57).
Migraine pain is present, although less frequently, in other mtDNA-associated diseases such as MERRF (myoclonus epilepsy with ragged red fibres), LHON (Leber's hereditary optic neuropathy) and NARP (neuropathy, ataxia and retinitis pigmentosa) (62–65).
Technical approaches that are actually being used to elucidate the mitochondrial genetic basis of migraine include the search for specific mtDNA mutations and association analysis (frequency of certain genetic markers in affected individuals vs. controls).
Search for specific mtDNA mutations
Molecular genetic studies in patients with different types of migraine have focused on mtDNA mutations that are strongly implicated in the mitochondrial encephalomyopathies, including the MELAS mutations (A3243G and T3271C), the MERRF mutation (G8344A), Kearns Sayre syndrome (KSS) common 4977 bp deletion, and the G3460A, T4160C and G11778A LHON mutations (17, 18, 24, 65–74)(Table 3). Most of these studies, even when performed in patients with a familial history of migraine, failed to detect any mtDNA mutations (Table 3).
Genetic studies in migraine
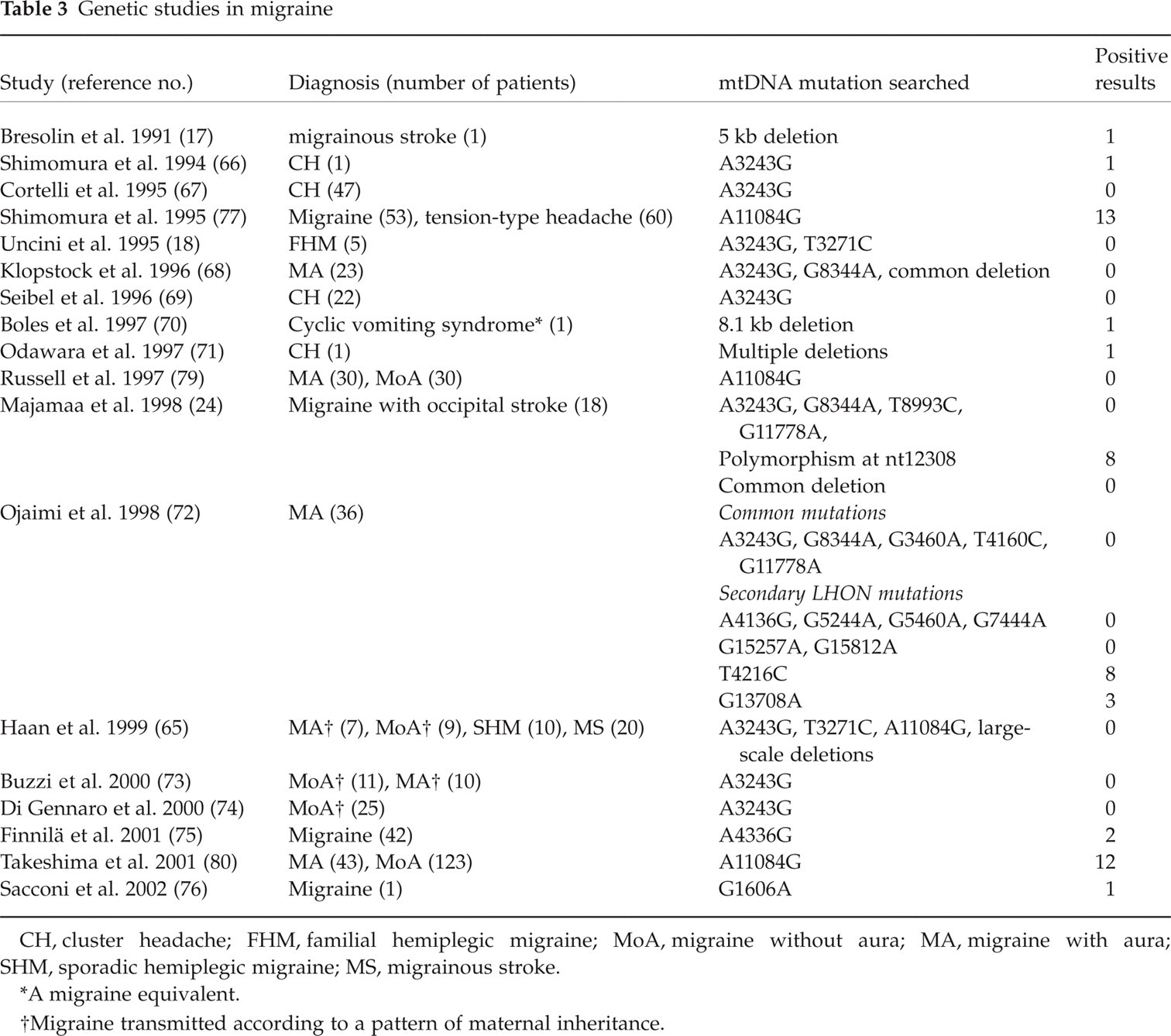
CH, cluster headache; FHM, familial hemiplegic migraine; MoA, migraine without aura; MA, migraine with aura; SHM, sporadic hemiplegic migraine; MS, migrainous stroke.
A migraine equivalent.
Migraine transmitted according to a pattern of maternal inheritance.
The few positive studies are limited to isolated migraineurs with mtDNA deletions (17, 70) or mtDNA point mutations (72, 75, 76) and to patients with cluster headache (CH) (66, 71) (Table 3). However, a common limitation of these studies is the clinical heterogeneity. According to Klopstock et al., cases reported in these studies usually ‘argue in favour of a syndrome beyond migraine’ (68).
Other apparently positive findings in patients with migraine have been subsequently challenged. Shimomura et al. found, in 13 out of 53 (24.5%) Japanese migraineurs, an A to G transition at mtDNA position 11084, in the coding region for the ND4 subunit of the mitochondrial respiratory chain (77). This mutation, first reported as a cause of MELAS (78), has not been found in Denmark (79) and has been demonstrated to be a common polymorphism in the Japanese population (80).
Several hypotheses may be raised to explain the negative results of most mitochondrial genetic searches in migraineurs:
Mutated mtDNAs might be absent or present in undetectable amount in blood samples. Mitochondrial diseases due to mtDNA defects, in fact, are heteroplasmic conditions, where variable degree of normal (wild-type) and mutated mtDNAs coexist in different cells or tissues (2, 5, 6). If the proportion of mutant genomes is expected to be low in oligosymptomatic patients, it would be more useful, although ethically and clinically inappropriate, to test the amount of mutated mtDNA in non-actively dividing tissues, such as brain parenchyma.
Migraine could be associated with still-unidentified mtDNA mutations.
Migraine might be caused by mutations in nuclear genes which may directly affect the energy production of the mitochondrial machinery (for example, genes encoding for subunits of the respiratory chain); additionally, nuclear genes may act indirectly, influencing the topographic expression of a mtDNA mutation in a specific neuronal population of the CNS (i.e. occipital cortex, where aura begins, brainstem structures that seem to be part of a neuronal network activated during a migraine attack, etc.) (81). As hypothesized for most mitochondrial encephalomyopathies, the selective vulnerability of specific neurons to a primary mtDNA mutation could be related to the activity of nuclear genes controlling the regulation of the threshold of normal mtDNA, required to complement mutant mtDNA among different cell types of the CNS (82–86). It could also be related to a mutation of nDNA-encoded factors involved in the post-transcriptional and/or post-translational regulation of mtDNA expression (86–98).
Association analysis
Association genetic analyses focus on the search for genetic markers that might be associated with migraine susceptibility, such as neutral polymorphisms, secondary mtDNA mutations, or nDNA mutations (48, 75). nDNA mutations, in particular, are mutations that have also become fixed in the population and that, interacting with nuclear or environmental factors, confer to the host an increased risk of developing the disease (75). Ojaimi et al. found a higher incidence of the secondary LHON mutations T4216C and G13708A in young stroke patients and in 36 MA patients in whom most common mtDNA mutations had been previously excluded (72). The authors hypothesized that the accumulation of so-called ‘minor’ mtDNA mutations might contribute to the pathogenesis of migraine and juvenile stroke even though, in this study, the frequency of the secondary LHON mutations in the MA group did not reach statistically significant levels (72).
Finnilä et al. recently demonstrated an increased risk of migraine in patients with the homoplasmic transition A to G at nt 4336 in the mtDNA tRNAGln gene (75). This mutation is normally present at low frequency in populations of European origin (75). Finally, Majamaa et al. analysed the mitochondrial haplotypes (normal population polymorphisms that seem not to induce any pathological variations in mitochondrial function) in 29 juvenile patients with occipital stroke (24). They found that 83% of the migraine stroke patients had the U mtDNA haplotype (as defined by a polymorphism at nucleotide 12308) and suggested that this mtDNA haplogroup may constitute a risk genome for migraine-associated stroke (24).
Indirect information from clinical trials
Riboflavin (vitamin B2), the precursor of several flavin nucleotides (i.e. flavin mononucleotide and flavin adenine dinucleotide) is required for the activity of flavoenzymes of the mitochondrial respiratory chain (89, 90). It has been shown to improve clinical and biochemical abnormalities in patients with MELAS and other mitochondrial diseases (91–94). In a placebo-controlled, double-blind trial, where patients in the active group received 400 mg/day of riboflavin, improvement in the primary and secondary endpoints was documented (95). The efficacy of riboflavin therapy seems to begin after 1 month and stabilizes after 3 months (95).
Coenzyme Q10, a small hydrophobic substance that acts as an electron carrier in the mitochondrial respiratory chain, has been recently investigated. In an open study, Rozen et al. treated 32 patients with a history of episodic migraine (with or without aura) at a dose of 150 mg/day, and found a reduced attack frequency and number of headache days per month (61.3% of patients had a >50% reduction in number of days with migraine headache) (96). In a recent and controlled study, Sandor et al. compared coenzyme Q10 (3 × 100 mg/day) and placebo in 42 migraine patients in a double-blind, randomized, placebo-controlled trial. Coenzyme Q10 was superior to placebo for attack frequency, headache days and days with nausea in the third treatment month. It was well tolerated. The 50% responder rate for attack frequency was 14.4% for placebo and 47.6% for coenzyme Q10 (number needed to treat: 3) (97).
From mitochondria to migraine
Migraine attacks seem to result from the interaction of intrinsic and environmental triggers in a susceptible brain (46, 81, 98–100). Interictal brain abnormal information processing is actually considered the biological basis of migraine (101) and has been demonstrated in migraineurs (especially in the occipital cortex) by many neurophysiological studies (98, 99, 102, 103). This abnormal information processing explains, at least partially, the efficacy of the anticonvulsant drugs (neuro-modulators) for migraine prophylaxis (104–106).
Along with other inherited factors, such as disturbances in magnesium metabolism and calcium channelopathies, an abnormality of mitochondrial oxidative metabolism increases neuronal excitability and reduces the threshold for triggering migraine attacks (46, 101). A mitochondrial defect may reduce the threshold for migraine attacks by several mechanisms:
Exogenous stimuli may, in cortical areas characterized by a reduced mitochondrial energy reserve, create an imbalance between neuronal energy supply and energy consumption that can accelerate the rise in lactate concentration normally occurring with stimulus-induced neuronal activity (positron emission tomography and nuclear magnetic resonance spectroscopy studies have demonstrated that photic stimulation normally induces an excess of glycolysis with a rise in lactate concentration in cerebral cortex) (103, 107–109). The local accumulation of lactate might further be increased by the coexistence in migraineurs of a habituation defect in sensory processing (alteration of a cortical adaptive mechanism characterized by reduction of the neuronal activity and decreasing lactate production that protect cerebral cortex against sensory overload) (103). The resulting imbalance of the brain metabolic homeostasis might activate the trigeminovascular system, triggering a migraine headache.
An impairment of oxidative metabolism confined to the trigeminal nerve nucleus or to brainstem structures, affecting somatosensory processing during a migraine attack, might locally enhance neuronal excitability. Recently, Cao et al. using blood oxygen level-dependent (BOLD) fMRI, have demonstrated that the activation of brainstem structures, such as the red nucleus and substantia nigra, precedes occipital cortex neurophysiological changes and clinical symptoms in visually triggered migraine attacks (110). Furthermore, a functional mitochondrial defect restricted to several brain structures, rather than affecting all the CNS, could be explained by the heteroplasmic nature of a disease due to a mtDNA mutation (primary mitochondrial defect) or by a focal magnesium deficiency causing an abnormality of mitochondrial oxidative phosphorylation (45, 101) (secondary mitochondrial defect).
The presence of mitochondrial abnormalities in the wall of meningeal blood vessels might explain the sensitivity of such structures to exogenous stimuli [i.e. nitric oxide (NO) donors, dietary triggers, etc.] in migraine sufferers (111). Furthermore, cerebral vascular abnormalities related to mitochondrial dysfunctions may account for variations in cerebral blood flow in some posterior regions associated with migraine aura, and attributed to cortical spreading depression (101, 112–115). It is noteworthy that pathological studies have widely demonstrated the presence of structurally abnormal mitochondria in the walls of cerebral and intramuscular small arteries of patients with MELAS (116–119).
Finally, it can be hypothesized that a mitochondrial dysfunction may be secondary to other primary biochemical dysfunctions. It is known that NO can induce headache in migraine patients and often triggers a delayed migraine attack (120, 121). The initial headache is thought to be caused via a direct action of the NO–cGMP pathway that causes vasodilation by vascular smooth muscle relaxation, while the delayed headache is likely to be a result of triggering trigeminovascular activation (121). Nitric oxide synthase (NOS) inhibitors are effective in the treatment of acute migraine (121). There is an increasing body of evidence demonstrating that NO, if produced in sufficient quantities, can elicit both reversible and irreversible inhibition of the mitochondrial electron transportation chain (122–125). In both astrocytes and neurons the inhibition of mitochondrial respiration by endogenously produced NO induces transient and modest decreases in cellular ATP concentrations. This mitochondrial impairment may serve as a cellular sensor of energy charges, hence modulating metabolic pathways, such as glycolysis, through AMP-activated protein kinase (AMPK) in astrocytes. In neurons, the NO derivative peroxynitrite anion triggers signalling pathways leading to glucose oxidation through the pentose–phosphate pathway to form reducing equivalents in the form of NADPH.
Conclusions
An impairment of oxidative metabolism may explain the ‘threshold character’ of migraine attacks. We have discussed morphological, biochemical and neuroradiological studies that may support the mitochondria core in migraine. Genetic evidence of mitochondrial involvement in this disease is less evident. Further studies are needed to clarify if migraine is still associated with unidentified mutations of the mtDNA or of nuclear genes which may equally affect the energy production of the mitochondrial machinery. Beyond the evident scientific interest of such studies, clinical consequences include eventual introduction of specific diagnostic tools for the clinician and the development of novel therapeutic approaches.