
Abstract
Although migraine is more common in women than men and often linked to the menstrual cycle, few studies have investigated the biological basis of hormonal influences on the trigeminovascular system. In the present study we investigated the effect of physiological levels (10-9 M) oestrogen on female rat trigeminal ganglia
Introduction
Migraine attacks are two to three times more prevalent in women than in men and are often associated with natural changes in levels of ovarian steroids during the menstrual cycle and at menopause (1, 2). Despite this strong link, few studies have examined the mechanism of ovarian steroid influences on neural systems relevant to migraine. Clinical data suggest that being female (and having high oestrogen levels) increases the probability of having migraine. Paradoxically, the sudden decreases in oestrogen levels at the time of menstruation increase the probability of having an attack. This apparent paradox suggests that oestrogen influences trigeminal pain by two independent mechanisms, i.e. chronic high oestrogen levels increase the likelihood of migraine by one mechanism, while sudden decreases in oestrogen activate a different mechanism. In the current study, we have addressed the first mechanism, whereby high oestrogen levels may increase the probability of having migraine (3). The trigeminal ganglia contain neurons innervating the head and face that carry pain information into the brain. These neurons also secrete neuropeptides capable of regulating blood flow and local inflammation in peripheral tissues such as meninges. Trigeminal innervation in rats is similar to human (4, 5), and rat models have been used to investigate several migraine-related events including cortical spreading depression (6), effects of triptans (7) and neurogenic vasodilation (8). Mitogen-activated protein kinases (MAPKs) are a family of intracellular signalling molecules that mediate signalling between extracellular events and cellular responses. The MAPK family includes extracellular signal-regulated protein kinase (ERK). Chronic ERK activation of sensory neurons occurs in experimental models of neuropathic and inflammatory pain, and treatment with ERK inhibitors alleviates pain-related behaviour (9, 10).
In several other reports in the literature, oestrogen has been shown to have antinociceptive effects in both male and female rats (11–13). By contrast, oestrogen may have pronociceptive effects on neuropathic pain after nerve constriction (14). Little information is available on potential effects of oestrogen on trigeminal neurons. To address this question, we investigated the influence of a physiological dose of oestrogen on gene expression and ERK activation using primary cultures derived from trigeminal ganglia of female rats.
Materials and methods
Animals
Random cycling female rats 50–60 days old were obtained from Harlan Breeding Laboratories (Indianapolis, IN, USA) and housed in the Laboratory Animal Resources Center at the University of Kansas Medical Center. Rats become sexually mature at 6 weeks, so at this age high levels of circulating oestrogen are present (15, 16). Rats were exposed to a 12 : 12-h light : dark schedule and provided with food and water
Isolation and culture of trigeminal ganglia
The culture procedure was modified from published procedures (17). For each experiment, trigeminal ganglia from 10 to 12 female rats were dissected from the bony base of the brain, rinsed in Hank's balanced salt solution (HBSS) and minced into small pieces with scissors. The minced tissue was then digested for 2 h in medium containing Leibovitz’ L15 with 1 mg/ml bovine serum albumin (BSA), 250 U/ml of CLSPA collagenase, 1 U/ml of ESL elastase, 5 U/ml of PAPL papain (enzymes from Worthington Biochemical Corp., Lakewood, NJ, USA). To the digest an equal volume of 30% Stractan (Larex, White Bear Lake, MN, USA) in Leibovitz’ L15 was added. The mixture was mixed by inversion, and then centrifuged for 10 min at 500
RNA preparation
TRIZOL was added to the cultures and cells were homogenized in TRIZOL Reagent (Life Technologies, Rockville, MD, USA). The total RNA was extracted as per the manufacturer's specifications, precipitated, and dissolved in RNase-free water. In the Affymetrix microarray system, we used the Rat Genome U34A chip. The Affymetrix GeneChip® Rat U34A chip contains 3192 full-length sequences, 3490 lesser annotated genes, and 649 EST clusters. Following hybridization, the analysis was performed at the Microarray Core Facility in the Biotechnology support facility at University of Kansas Medical Center. Briefly, following RNA isolation the first-strand synthesis was performed using 10 µg of total RNA, a T7-(dT)24 oligomer with the Superscript Choice System (Invitrogen, Carlsbad, CA, USA). The T7 promoter introduced during the first-strand cDNA synthesis was then used to direct the synthesis of cRNA by using T7 RNA polymerase (Enzodiagnostics, Farmingdale, NY, USA) and biotinylated deoxyribonucleotide triphosphates. The biotinylated cRNA was fragmented to a mean size of 200 bp before hybridization. Total RNA and biotin-labelled cRNA were tested for integrity and size by resolving on Agilent RNA 6000 Nano Laboratory Chips (Agilent, Palo Alto, CA, USA). Hybridization was performed at 45°C for 16 h [0.1
Gene chip data analysis
Data were analysed with Affymetrix Microarray suite MAS 5.0 software, which used the one-sided Wilcoxon's signed rank test to generate a detection
Up-regulated genes

RNA from control and 17β-oestradiol-treated cell cultures were hybridized on Affymetrix Rat RG_U34A gene chips. Three independent sets of experiments were performed. Each set consisted of trigeminal ganglia culture prepared from 10 female rats using six chips in total, three for control, three for experimental groups. In the treated group the cells received 10−9
Kinase screen
The effect of 17β-oestradiol on various kinases was assessed through the use of multi-immunoblot phosphokinase screen KPSS 3.1 performed by Kinexus Bioinformatics Corp. (Vancouver, BC, Canada). For this screen, cell lysates were prepared from cultured trigeminal ganglia. In brief, the media was removed from the wells, washed with PBS followed by addition of lysis buffer consisting of Tris–HCl buffer (pH 7.4) containing 1% Triton X-100, 10% glycerol. To the lysis buffer were added protease inhibitors at the final concentration 30 m
At least three to four wells were pooled to generate one sample. The mixture was centrifuged at 10 000
Western blot analysis
Cell lysates were prepared from control and oestrogen-treated cell cultures as mentioned above. The samples containing 7.5 µg protein were separated in 10% SDS–polyacrylamide gels and electrophoretically transferred to PVDF membranes. Membranes were incubated in blocking buffer containing 4% BSA in TBS with 0.1% Tween 20. Membranes were probed with rabbit anti-ERK 1 and 2[pTpY185/187] phospho and anti-ERK 1 and 2 pan antibody (Biosource, Camarillo, CA, USA) in blocking buffer at room temperature for 3 h at 1 : 400 and 1 : 3000 dilution, respectively. The membrane was washed three times in TBS–T and then detected with a biotinylated anti-rabbit secondary antibody. The Vectastain ABC-AmP kit from Vector Laboratories was used for Western blot immunodetection. The blot was scanned and imaged using a BioRad ChemiDoc XRS system.
Immunocytochemistry
The cultures were fixed with chilled methanol and were maintained at − 20°C for 10 min followed by washing with PBS. The cells were permeabilized with 0. 2% Triton X-100 and blocked with Cyto Q background buster from Innovex Biosciences (El Cajon, CA, USA). Cultured neurons were stained overnight at room temperature with rabbit anti-ERK 1 and 2[pTpY185/187] phosphospecific antibody (1 : 100; Biosource International), rabbit anti-peripherin (1 : 50; SantaCruz Biotechnology, Santa Cruz, CA, USA), a marker of nociceptive sensory neurons with unmyelinated axons (23) or rabbit anti-oestrogen receptor-alpha (ER-α) (1 : 50; Santa Cruz Biotechnology). After four washes with PBS, the fluorescent secondary antibody Texas Red-conjugated sheep anti-rabbit from Jackson ImmunoResearch Laboratories (West Grove, PA, USA) was used at 1 : 1000 dilutions for 30 min at room temperature followed by counterstaining in 4′-6-diamidino-2-phenylindole (DAPI) for 5 min. Slides were washed and coverslipped with slow fade antifade media (Molecular Probes, Eugene, OR, USA). Controls consisted of omitting the primary antibody from the reaction, and no staining was observed in control slides. Sections were observed on a Nikon (Garden City, NY, USA) Eclipse TE300 microscope equipped with a Spot Digital Camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) and Zeiss LSM 510 confocal microscope.
Results
First, we demonstrated that trigeminal neurons contain ER-α. In the cultured trigeminal neurons from female rats, ER-α immunoreactivity was present in a cytoplasmic location including neurites (Fig. 1a).

(a) Oestrogen receptor-alpha (ER-α) immunocytochemistry. Trigeminal ganglia cultures were grown in Falcon eight-chambered culture slides. The culture was immunostained with rabbit anti-ER-α antibody and sheep anti-rabbit Texas Red-conjugated secondary antibody. ER-α immunoreactivity is present in neuronal cytoplasm. The cells have been counterstained with 4′-6-diamidino-2-phenylindole (DAPI). Scale bar = 25 µm. (b) Phospho ERK 1/2 immunocytochemistry. Trigeminal ganglia cultures were grown in Falcon eight-chambered culture slides. The culture was immunostained with rabbit Phospho ERK1/2 antibody and goat peripherin antibody. Peripherin is a marker of sensory neurons with unmyelinated axons, i.e. unmyelinated nociceptors. Sheep anti-rabbit Texas Red and fluorescein-conjugated anti-goat secondary antibodies were used followed by counterstaining of cultures with DAPI. Photomicrographs were taken using an LSM 510 confocal microscope (Carl Zeiss). Scale bar = 10 µm.
Microarray analysis comparing oestrogen-treated with vehicle-treated trigeminal neurons revealed several genes that were consistently up- or down-regulated in response to the oestrogen treatment. Genes responding to oestrogen treatment induced by 1.4-fold or greater (Table 1) included MHC-1b, activity and neurotransmitter-induced early gene 7 (ania7), synapsin-2, phosphoserine aminotranferase, endothelin receptor type B, and ERK-2. Genes responding to oestrogen treatment by down-regulation by 1.4-fold or greater (Table 2) included interleukin-1β receptor 1 (IL-R1), bradykinin B2 receptor, the β chemokine CCL20, N-tropomodulin, a GABA transporter protein, fetal intestinal lactase-phlorizin hydrolase, carcinoembryonic antigen-related protein, zinc finger protein 36, epsin 1 and cysteine string protein.
Protein kinase arrays and Western blots confirmed effects of oestrogen treatment on ERK kinases (Fig. 2). We observed that in response to these physiological doses of oestrogen both phosphorylated ERK1 and MEK1, another enzyme from the MAPK kinase signalling cascade, were elevated. Quantification of these data indicated a mean increase in phosphoERK by 1.4-fold in three experiments.
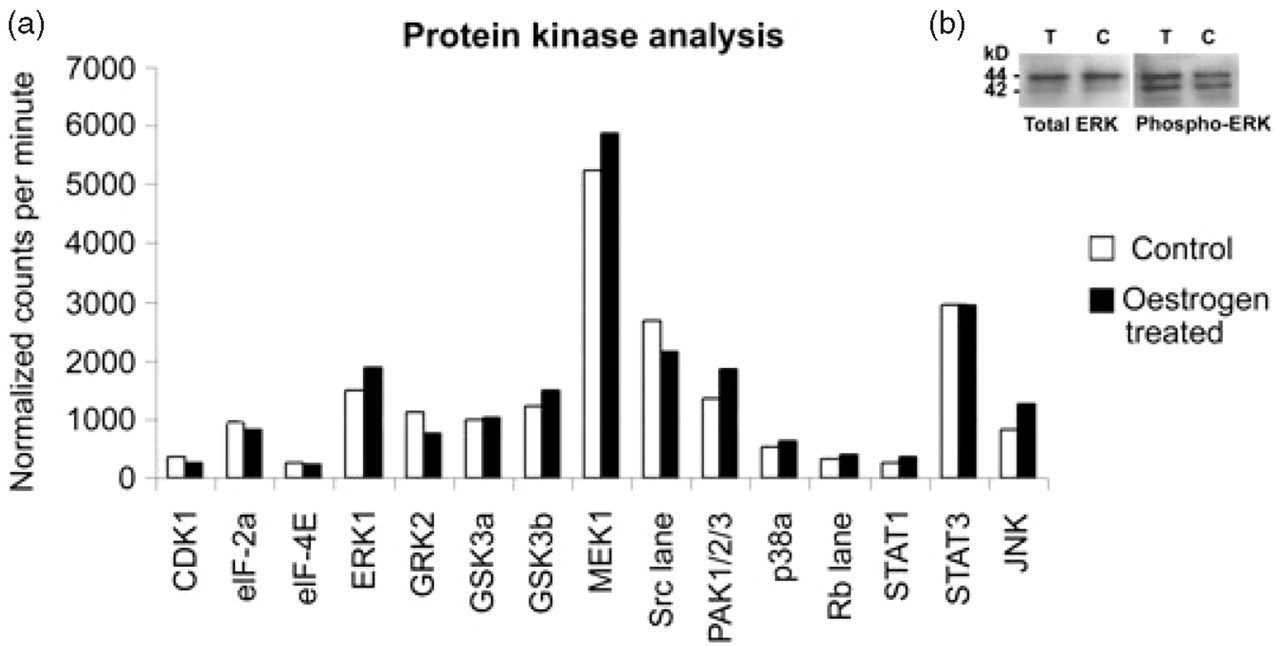
(a) Protein kinase analysis: Kinetworks KPSS-3.1 phosphoprotein analysis of rat trigeminal ganglia cultures. The proteins listed are represented in terms of fold change compared with control. The proteins listed here show an increase in ERK1 and upstream kinase MEK1. (b) Lower panel shows the Western blot analysis of trigeminal ganglia cultures with total ERK1/2 and phosphoERK1/2 antibodies. Each of the lanes was loaded with 7.5 µg of protein. The bands were detected using chemiluminiscence in the BioRad ChemiDoc XRS system. p44 corresponds to ERK1 and p42 corresponds to ERK2. There was 1.5-fold increase in the band intensity of phosphoERK1 of treated sample (▪) compared with the control (□).
Immunocytochemistry confirmed the localization of activated ERK in nociceptors (Fig. 1b). Figure 1b demonstrates phosphoERK immunoreactivity (red) in neurons expressing peripherin (green). Overlap of these two proteins is seen as yellow fluorescence. Nuclei are stained blue with DAPI. Expression of peripherin confirms the neuronal identity of the cells as well as demonstrating the cells expressing phosphoERK as primary afferent nociceptors (23, 24).
Discussion
Our study demonstrates that oestrogen has direct effects on gene expression and intracellular signalling in cultured female rat trigeminal neurons, which may be important in hormonal regulation of pain systems. Physiological doses of oestrogen up-regulate genes involved in synaptic transmission and intracellular signalling and down-regulate bradykinin and IL receptors. This study addresses how chronic high oestrogen levels increase the likelihood of migraine rather than how sudden decreases in oestrogen precipitate attacks. The present work addresses the question of direct effects of oestrogen on trigeminal neurons but has important limitations. In order to grow trigeminal neurons in culture, they must be axotomized. After 3 days, when the oestrogen treatment was initiated, the neurons have regrown their processes. Possible effects on the phenotype due to axotomy or the culturing process are currently unknown.
We demonstrated the presence of ER-α in female rat trigeminal neurons in a cytoplasmic distribution and in neurites. Cytoplasmic ERs function by activating transcription at AP-1 sites in response to 17β-oestradiol (25), by inducing activation of STAT related promoters (26), and by inducing phorphorylation of ERK (27). ER-α in neurites mediates rapid effects of oestrogen, including local activation of ERK (28). All of these mechanisms are potentially involved in regulating expression of the genes demonstrated by our microarray analysis.
Microarray analysis demonstrated that oestrogen treatment up-regulated expression of ERK-2 mRNA, and protein analysis demonstrated that oestrogen treatment increased ERK activation. ERKs are MAPKs that are activated by membrane depolarization, calcium influx, mitogens and, in the central nervous system, by neurotrophins and neurotransmitters (29). ERK activation contributes to nociception as activation of nociceptive fibres induces phosphoERK (pERK) in sensory neurons in an intensity-dependent manner, and ERK antagonists inhibit capsaicin-induced hyperalgesia (30). ERK activation also contributes to inflammatory pain (13, 30, 31). The ERK pathway is necessary for capsaicin- and NGF-induced heat hyperalgesia (32). Inflammation increases pERK in nociceptive DRG neurons, and treatment with the MAPK kinase inhibitor U0126 decreases thermal hyperalgesia after capsaicin treatment (30). ERK activation has been used as a marker for the functional activation of neurons in a model of peripheral inflammation (33). Axotomy, a model of neuropathic pain, induces ERK in medium and large DRG neurons and satellite cells. Inhibiting ERK activation with U0126 also decreases pain related-behaviour after axotomy. ERK activation increases neuron excitability by phosphorylating the Kv4.2 channel, which mediates a transient outward potassium current (34). Phosphorylation inhibits that channel, resulting in increased excitation of the neuron. Thus, activation of nociceptors activates ERK, as do inflammatory and neuropathic pain, and ERK activation increases neuron excitability. Our data suggest that physiological levels of oestrogen regulate the Raf1→MEK1/2→ERK1/2 signalling pathway in trigeminal ganglia.
Other genes revealed by the microarray analysis may be involved in regulating biochemical, structural, and electrical properties of trigeminal neurons, or their responses to nerve injury or tissue inflammation. Oestrogen treatment up-regulated class 1b MHC mRNA in trigeminal neurons. Class I MHC is a glycoprotein, expressed by most cells, that is involved in antigen presentation. Adult neurons express little class I MHC, but both injury and inflammation up-regulate it. MHC-I is up-regulated in motor neurons after axotomy (35, 36) and in hypothalamic neurons in both chronic and acute inflammation models (37). Class I MHC expression also increases in response to neural activity (38). Increased class I MHC expression in response to chronic infection and acute inflammation may be involved in antigen presentation of infiltrating T cells (39), in presentation of self antigens (40), and possibly to mark injured neurons (41) and damaged axons (42). Sensory neurons normally do not express class I MHC because they express high levels of suppressor of cytokine signalling-1 (SOCS1), which down-regulates class I MHC (43). The mechanism whereby oestrogen up-regulates class I MHC is unknown, but one possibility is that oestrogen down-regulates SOCS1 expression.
Oestrogen treatment up-regulated endothelin receptor B and synapsin 2, which may be involved in increased excitability after injury. The peptide endothelin is involved in nociceptive signalling independently of its vasoconstrictive effects. Tissue endothelin levels increase after cutaneous injury, where it is thought to contribute to local inflammation (44) and pain (45–47). Activation of the endothelin B receptor inhibits endothelin-induced nociceptive behaviour (48). Thus, oestrogen treatment also up-regulates a gene that may be antinociceptive. Up-regulation of synapsin 2 suggests that oestrogen treatment may alter excitability by increasing neurotransmitter release. Synapsin 2 is a major phosphoprotein found in nerve terminals, where it is shown to be associated with maintenance of synaptic vesicle contact with actin filaments (49, 50). Physiological stimuli, including pregnancy and lactation, up-regulate synapsin 2 expression in the hypothalamus (51).
Oestrogen treatment also up-regulated two other genes: activity and neurotransmitter-induced early gene 7 (ania-7) and phosphoserine aminotransferase. The potential functions of these two genes in sensory neurons are unknown.
Oestrogen treatment down-regulated several genes which are potentially relevant to calcitonin gene-related peptide (CGRP) release from nociceptors. Down-regulation of bradykinin B2 receptor suggests that this receptor is expressed at highest levels during low oestrogen phases of the cycle. Bradykinin induces release of CGRP from sensory neurons (52, 53), which should be greatest when oestrogen levels are low. Down-regulation of the IL-1β receptor may have similar effects, as IL-1 receptor is expressed in sensory neurons (54) and IL-1β also induces release of CGRP from sensory neurons (55). This would provide an additional mechanism to increase CGRP release from sensory neurons at low oestrogen phases of the cycle.
Oestrogen down-regulated several other genes with potential relevance to migraine, including zinc finger protein 36, fetal intestinal lactase-phlorizin hydrolase, N-tropomodulin, epsin 1 and cysteine string protein. Zinc finger protein 36 has an anti-inflammatory function achieved by binding-3′-untranslated regions of the mRNAs encoding tumour necrosis factor (TNF) leading to accelerated mRNA degradation (56). Oestrogen down-regulates fetal intestinal lactase-phlorizin hydrolase (LPH), which is involved in vitamin B6 release (57). Interestingly, vitamin B6 pyridoxine deficiency causes a sensory neuropathy, while pyridoxine overdoses are neurotoxic to large fibre sensory neurons. N-Tropomodulin is a neuron-specific form of tropomyosin binding protein (58). Tropomodulins regulate the length of actin filaments and play a role in synaptic function, providing another potential mechanism for hormones to alter excitability (59). Cysteine string protein (CSP) is an abundant synaptic vesicle protein demonstrated to be necessary for continued synaptic function (60). Epsin 1 is also involved in synaptic function (61). Down-regulation of CSP and epsin 1 provide additional potential mechanisms for oestrogen to regulate neuronal excitability.
Oestrogen treatment down-regulated other genes involved in inflammation and excitability. CCL20/MIP3α/ST38 is a member of the CC chemokine family that attracts immature dendritic cells, effector/memory T cells and B cells to injury sites (62). CCL20 is up-regulated in ischaemic brain and in models of brain inflammation (63). Oestrogen treatment also down-regulated GABA transporter GAT3. GABA immunoreactive neurons are present in trigeminal ganglia (64, 65). GABA B receptor agonists reduce excitability of trigeminal neurons (66), suggesting that trigeminal GABA may regulate trigeminal excitability.
This study demonstrates that oestrogen treatment alters expression of genes relevant to migraine in trigeminal neurons
Footnotes
Acknowledgements
Supported by GlaxoSmithKline and NIH grant DE01582. We thank Paul Strijbos for comments on the manuscript.