
Abstract
The present work aimed 1) to evaluate whether an increase in galanin or galanin receptors could be induced in the nucleus basalis magnocellularis (nbm) by degeneration of the basalocortical neurons from the cortex and 2) to analyze the consequences of such an increase on cortical activity. First, a mild ischemic insult to the frontoparietal cortex was performed to induce the degeneration of the basalocortical system; galanin immunoreactivity, galanin binding sites, and cholinergic muscarinic receptors were quantified through immunocytochemistry and autoradiography. Second, galanin infusions in the nbm were undertaken to mimic a local increase of the galaninergic innervation; cortical acetylcholine release, cerebral glucose use, and cerebral blood flow were then measured as indices of cortical activity. As a result of the cortical ischemic lesion, the postsynaptic M1 and presynaptic M2 muscarinic receptors were found to be reduced in the altered cortex. In contrast, galaninergic binding capacity and fiber density were found to be increased in the ipsilateral nbm in parallel with a local decrease in the cholinergic markers such as the muscarinic M1 receptor density. Galanin infusion into the nbm inhibited the cortical acetylcholine release and cerebral blood flow increases elicited by the activation of the cholinergic basalocortical system but failed to affect acetylcholine release, cerebral blood flow, and cerebral glucose use when injected alone in the nbm. These results demonstrate that degeneration of the basalocortical system from the cortex induces an increase in galaninergic markers in the nbm, a result that might suggest that the galaninergic overexpression described in the basal forebrain of patients with Alzheimer's disease can result from a degeneration of the cholinergic basalocortical system from the cortex. Because galanin was found to reduce the activity of the basalocortical cholinergic system only when this one is activated, galanin might exert its role rather during activation deficits than under resting conditions such as the resting cortical hypometabolism, which is characteristic of Alzheimer's disease.
Keywords
Galanin, a 29 amino-acid neuropeptide widely distributed throughout the peripheral and central nervous systems, is involved in neurodegenerative and regenerative responses (Holmes et al., 2000). In Alzheimer's disease (AD), galanin fibers have been reported to hyperinnervate the remaining cholinergic neurons of the nucleus basalis of Meynert (NBM) (Bowser et al., 1997;Chan-Palay, 1988). The origin of this overexpression or increase in innervation in AD is, however, unknown. If galanin has been extensively reported to be overexpressed after neuronal damage (Cortés et al., 1990;de Lacalle et al., 1997), the mechanisms that lead to the galaninergic overexpression in the NBM are unknown. Several studies have reported local galaninergic overexpression after lesion of the nucleus basalis magnocellularis (nbm, the equivalent of the primate NBM) (Unger and Schmidt, 1993) or of the horizontal limb of the diagonal band of Broca (de Lacalle et al., 1997). These studies suggest that this hyperinnervation may result from the local degeneration of the cholinergic neurons (Hartonian et al., 2002). However, the degeneration of cholinergic pathways in AD is likely to be secondary to the degeneration of cortical areas (Henderson, 1996). The first aim of this work was thus to determine whether a galaninergic hyperinnervation in the rat nbm occurs after damage of the basalocortical system from the cortex. This question has been addressed through the induction of a mild, controlled, ischemic insult to the frontoparietal cortex, hypothesized to induce a degeneration of the basalocortical pathway (Kataoka et al., 1991;Liberini et al., 1994), and the assessment of galanin immunoreactivity and binding site density within the nbm, in parallel with the quantitation of cholinergic markers.
The putative functional consequences of such a galaninergic hyperinnervation in NBM in AD are also unknown, although hypothesized to induce an exacerbation of the cholinergic deficit and, putatively in turn, an exacerbation of the deficit in cortical activity. Indeed, galanin has been reported to be functionally involved in the rat basal forebrain where it exerts an inhibitory modulation upon the cholinergic septohippocampic and basalocortical systems (Barbelivien et al., 1995, 1998;Fisone et al., 1987). Patients with AD are characterized by early decreases in cerebral glucose use (CGU) and cerebral blood flow (CBF) in temporoparietal and frontal cortices (Friedland et al., 1983;Prohovnik et al., 1988). Although the mechanisms for these alterations are unknown, functional and anatomic data suggest that these alterations are not merely the reflection of a loss of tissue, such as demonstrated by Salmon et al. (1996). Damage to the NBM is among the most severe and consistent features of AD (Perry et al., 1982), and the hypometabolism/hypoperfusion characteristics of AD have been at least partly attributed to the cholinergic deafferentation disrupting neocortical synaptic activity (Geaney et al., 1990). Indeed, similar patterns of decline in neocortical choline acetyltranferase (ChAT) and CGU have been observed in AD (Friedland et al., 1983;Procter et al., 1988), and acetylcholinesterase inhibitor treatments result in a reduction of the hypometabolism and hypoperfusion (Nordberg, 1993). Perry et al. (1982) demonstrated, however, that the extensive reduction in cortical ChAT activity in AD is not directly reflected by the neuronal loss in the NBM, suggesting the existence of alterations that lead to a decrease in the local production of acetylcholine (ACh). One might thus hypothesize that the cortical hypometabolism and hypoperfusion in AD result from the combined effect of the neuronal loss in the NBM and another process that decrease the activity of the surviving basalocortical cholinergic neurons. The galaninergic hyperinnervation of the cholinergic neurons of the NBM in AD (Bowser et al., 1997;Chan-Palay, 1988) could represent such a key. The second aim of this work was thus to test the hypothesis that the increase in galanin in the NBM can reduce cortical neuronal activity. To this end, indices of cortical activity such as CGU, CBF, and cortical ACh release were measured after galanin injections into the nbm, hypothesized to mimic an increase in galanin innervation in this brain region.
MATERIALS AND METHODS
Animals
Experiments were performed upon male Sprague Dawley rats (approximately 250 g; Centre d’Elevage René Janvier, France), maintained on a 12-hour light/dark cycle and allowed food and water ad libitum in thermoregulated (22 ± 2°C) and humidity-controlled (55 ± 10%) animal facilities. The rats used for CGU measurements were fasted for 18 hours before the study. All experimental protocols were undertaken in compliance with the European Community Directives and the French law on animal experimentation (personal authorization No. 14–05 for F.D.).
Experiment 1: Lesion of the basalocortical system from the cortex and effect upon the galaninergic system in the nbm
Lesion of the basalocortical system through a mild ischemic insult to the cortex.
Based upon the high density of cholinergic fibers innervating the frontoparietal cortices from the nbm and the known effect of neocortical infarction on damage to the neurons of the nbm (Figueiredo et al., 1993;Kataoka et al., 1991;Liberini et al., 1994), a controlled ischemic insult to the frontoparietal cortex was performed to induce a lesion of the basalocortical pathway. This was achieved by a distal, unilateral, occlusion of the middle cerebral artery (MCA) of rats under halothane anesthesia and thermoregulation (37.5 ± 0.5°C) (Tamura et al., 1981). In brief, a scalp incision was made between the eye and the ear of each rat, the zygomatic arch was removed, and the temporal muscle was incised to expose the cranial bone. A small craniotomy (2 mm diameter) was carried out with a high-speed drill close to the foramen ovale; the dura was incised and the MCA was occluded by a bipolar electrocoagulation (0.7–1.3 μA, 5–8 seconds). Stopping of the blood flow was checked; tissues were then closely sutured and animals (n = 18) maintained at 37.5°C for 1 hour before being placed back into their cages. Sham-operated animals (n = 16) were undertaken in parallel with similar procedures except that the MCA were not coagulated.
Eighteen days after surgery, rats were used either for immunocytochemical analysis of the galanin expression or for autoradiographic quantitation of galanin binding sites (Gal-R) and muscarinic receptors. Rats used for immunocytochemistry were intracardiacally perfused under anesthesia with 50 mL of cold heparinized saline and 500 mL 2% paraformaldehyde-15% picric acid phosphate buffer; brains were dissected out, cryoprotected for 24 hours in the same buffer containing 20% sucrose, blocked into embedding medium, frozen on dry ice, and then maintained at −80°C. Animals used for autoradiography were subjected to euthanasia; then, the brains were rapidly excised, frozen in cold isopentane (−45°C), and stored at −80°C.
Immunocytochemical determination of galaninergic fibers.
Frozen brains were transferred to a cryomicrotome (−20°C) to obtain adjacent 12-μm coronal sections from AP +7.2 mm to AP +11.7 mm (anteroposterior distances from the interaural line) (Paxinos and Watson, 1986). Sections were mounted on microscopic slides, incubated in Coons buffer (2 × 5 minutes), and then incubated for 18 hours at room temperature in the primary antibody solution (rabbit anti-GAL, 1/800 in Coons buffer containing 0.25% Triton X100, G. Tramu, Talence, France) (Anglade et al., 1994). After three rinses in fresh buffer, sections were incubated for 90 minutes in the presence of the secondary antibody (donkey anti-rabbit, 1/400 in Coons buffer with 0.25% Triton X100, Interchim, France) labeled with fluorescein isothiocynate (FITC). Sections were mounted under coverslips and analyzed under epifluorescence. Changes in immunolabeling were studied on digitalized images taken from the ipsi- and contralateral nbm at three anteroposterior levels (AP +7.20, +7.70, and +8.20) (Paxinos and Watson, 1986); the density of galanin immunoreactive fibers was quantified as the surface (μm2) corresponding to immunolabeling in a fixed-sized region (0.125 mm2; Image J, NIH, U.S.A.). Labeling specificity was assessed on parallel sections by the determination of the extinction of the immunoreactive signal through the use of increasing concentrations of galanin (10−8 to 10−2 M) coincubated with the antibody.
Autoradiographic determination of galaninergic and muscarinic receptors.
Eight sets of adjacent coronal sections (14 μm) were obtained at −20°C from the frozen brains: seven sets corresponding of 12 sections taken at AP +11.70, +10.60, +9.48, +7.20, +5.86, +4.20, +3.20, +2.20, +1.36, +0.28, −0.68, and −1.04 mm (Paxinos and Watson, 1986) for autoradiography and histologic counterstaining, and one set of 12 equidistant sections that cover the entire infarct for the determination of the infarct volume. Six of the seven sets were dedicated to the receptor binding study and thus stored at −80°C until the day of the autoradiographic experiments, whereas the last set was maintained at 4°C until histologic staining (cresyl violet).
Galaninergic binding sites.
Two adjacent sets of sections were incubated twice (2 × 10 min) in 50mM Tris-HCl (pH 7.4, 25°C) containing 250 mM sucrose, 2 g/L bovine serum albumin (BSA), and 0.02 g/L bacitracine (Dutriez et al., 1996). Then, sections were immersed for 120 minutes in the same buffer in the presence of 0.3 nM [125I]galanin (2,200 Ci/mmol, NEN Life Sciences, France) supplemented (nonspecific binding) or not (total binding) with 1 μM galanin. Slides were then rinsed in cold 50 mM Tris-HCl (2 × 5 min, 4°C) and dried under a cold stream of air. Sections were then exposed for 6 days to autoradiographic films ([3H]Hyperfilms, Amersham, France) in parallel with [125I]microscales (Amersham, France).
M1 and M2 muscarinic binding sites.
Four sets of sections were incubated for 15 minutes at room temperature in a Krebs buffer (120 mM NaCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 5.6 mM glucose, 25 mM NaHCO3, 2.5 mM CaCl2, 4.7 mM KCl, pH 7.4) (Le Jeune et al., 1995). Then, sections were incubated for 60 minutes in the same buffer in the presence of 3 nM [3H]pirenzepine (M1 muscarinic receptors, 77.9 Ci/mmol, NEN Life Sciences, France) (Le Jeune et al., 1995) or 2 nM [3H]AF-DX 384 (M2 muscarinic receptors, 133.1 Ci/mmol, NEN Life Sciences, France) (Aubert et al., 1992), with (non-specific binding) or without (total binding) 1 μM atropine. Slides were then rinsed in cold 50 mM Tris-HCl buffer (3 × 4 minutes, pH 7.4, 4°C), dipped in cold distilled water, and dried under a cold stream of air. Sections were then exposed for 28 ([3H]pirenzepine) or 14 days ([3H]AF-DX 384) to Hyperfilms in parallel with [3H]microscales (Amersham, France).
Quantitative analysis of autoradiographic films.
After development and fixation, films were analyzed using densitometry (Biocom Rag 200); binding densities (fmol/mg protein) in selected regions of interest (determined on cresyl violet stained adjacent sections) were obtained by transforming gray levels to radioactive concentrations according to the best-fit relationships determined on data from the coexposed radioactive standards.
Histologic staining and determination of infarct volumes.
One set of brain slices covering the infarcted region was stained with eosinehemateine, dehydrated, and mounted under cover-slips. Infarct volumes (mm3) were quantified by integration of the necrotic surface (defined by the relative paleness of the staining) on each section as a function of the distance between sections (Biocom Rag 200).
Experiment 2: Functional effects of galanin injections in the nbm of the rat
Surgery.
Rats were anesthetized with urethane (Sigma, France) (500 mg·kg−1 intraperitoneally) and α-chloralose (Sigma, France) (50 mg·kg−1 second) after induction with halothane (4.5% then 1% in O2) (see for details Barbelivien et al., 1999). Catheters were placed bilaterally in the femoral veins and arteries, and the trachea was cannulated. The skin incisions were infiltrated with xylocaine and sutured. The rats were ventilated artificially (Harvard Apparatus, Model 683) and transferred into a stereotaxic frame (David Kopf Instruments, Model 1430). Continuous monitoring of arterial pressure and heart rate was recorded through one of the femoral catheters connected to a pressure transducer and amplifier (Gould, Model 1170). After craniotomy, a stainless steel cannula (200 μm outer diameter; for CBF-CGU coupling experiments) or a glass micropipette (50 μm outer diameter at the tip; for microdialysis experiments) was inserted into the right nbm (interaural axis 7.2 mm, lateral 2.9 mm, and −8.2 mm from the surface of the calvarium) (Paxinos and Watson, 1986). After completion of the surgical procedures, halothane was discontinued. Arterial blood was sampled periodically for measurements of pCO2, pO2, and pH; tidal volume was adjusted to maintain values of pCO2 35–39 mm Hg, pO2 > 150 mm Hg, and pH 7.4. Rectal temperature was maintained constant (37°C) by means of a thermoregulated blanket.
Measurements of CGU and CBF.
Intracerebral microinjections.
The stainless steel cannula was connected to a microinfusion pump (Carnegie Medicin, CMA/100). Each animal received 500 nL of 0.9% NaCl (saline; n = 8) or galanin (Bachem, Switerzland) (315 pmoles) (galanin; n = 7), infused over a period of 50 minutes.
CGU and CBF measurements.
CGU and CBF were measured autoradiographically in the same animal through the use of a two-tracer technique with [14C]iodoantipyrine (IAP) for CBF and [18F]fluorodeoxyglucose (FDG) for CGU (Barbelivien et al., 1999).
Two hours after the administration of urethane and α-chloralose, and 5 minutes after the onset of the intracerebral microinjection, a tracer dose of [18F]FDG (130 ± 18 MBq; mean ± SD) were injected intravenously as a bolus. After injection of [18F]FDG, and over the subsequent 45 minutes, arterial samples were collected at predefined time intervals and centrifuged to measure the plasma concentration of [18F]FDG (Packard, COBRA) and glycemia (Beckman, Glucose analyzer 2). Forty-four minutes after [18F]FDG injection, [14C]IAP (1.11 MBq dissolved in 0.7 mL 0.9% NaCl) (NEN, France) was perfused intravenously at a constant rate of 15 μL·s−1 (Harvard Apparatus, Model 11) and arterial samples were collected every 3 to 4 seconds. Approximately 35 seconds after the onset of IAP perfusion, euthanasia was performed by a bolus injection of thiopental (200 mg·kg−1), followed by decapitation. The brains were removed rapidly, frozen in isopentane (−45°C), and serial coronal brain slices (20 μm) were obtained immediately with a cryomicrotome. At the level of the nbm, sections were collected and stained (Cresyl violet) to ascertain that the cannula tip lay within the nbm (Paxinos and Watson, 1986). Sections for autoradiographic analysis were mounted on coverslips and dried rapidly on a hot plate (60°C). Thereafter, sections and [18F] standards, prepared in parallel, were exposed to x-ray films (Kodak, X-OMAT-AR) to obtain the tissue concentrations of [18F]FDG; to rule out a putative cross contamination with [14C]IAP, the time of exposure was always less than 2 hours. After complete [18F] decay (3 days later), the same sections and calibrated [14C] standards (ARC, 146-C) were exposed to x-ray films for 5 days to obtain tissue concentrations of [14C]IAP. The plasma concentrations of [14C]IAP were determined subsequently (Packard TRI-CARB 2200).
CBF and CGU calculations.
Autoradiograms were quantified through the use of a computerized image analysis system (Biocom Rag 200) according to Sokoloff et al. (1977) for CGU (μmol·100 g−1·min−1) and Sakurada et al. (1978) for CBF (mL·100 g−1·min−1). For each region of interest, readings for CGU and CBF were averaged independently for ipsi- and contralateral hemispheres.
Measurement of cortical ACh release.
Cortical ACh release and CBF were measured in the same animal through the use of a brain microdialysis probe and [14C]IAP, respectively. Based upon our previous studies (Barbelivien et al., 1995, 1998), which demonstrate that galanin exerts a modulatory inhibitory action rather than an direct effect upon the cholinergic basalocortical system, the effects of galanin infusions into the nbm upon cortical ACh release was investigated in either basal or cholinergically-stimulated conditions (Barbelivien et al., 1998). Cortical ACh release was measured in the frontoparietal cortex because this cortical region receives a dense cholinergic innervation from the nbm.
Intracerebral microinjections.
The glass micropipette was connected to a nanoinfusion pump (WPI, Nanopump A1400). Each animal received a microinjection (100 nL over 10 minutes) of 0.9% NaCl (n = 6, saline group), 50 nmol of carbamylcholine chloride (carbachol) (Sigma, France) (n = 6, carbachol group), a mixture of 50 nmoles of carbachol and 63 pmol of galanin (n = 5, coinjection group), or 63 pmol of galanin alone (n = 6, galanin group) (Bachem, Switzerland). Localization of the infusion sites was confirmed during brain microdissection.
Intracerebral microdialysis.
Under halothane anesthesia (1.5%), a small craniotomy was performed, and a dialysis probe (300 μm outer diameter, 4 mm length, 4,400 Da) was tangentially implanted into the right parietal cortex. A modified Ringer's solution (145 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 1.0 mM MgCl2, pH 7.4) containing 0.5 μM neostigmine (Sigma, France) was perfused (2 μL·min−1) through the probe with a microinjection pump (CMA/100). Fifteen minutes after the insertion of the microdialysis probe, the rats received an injection of the anesthetic agents (urethane 500 mg/kg intraperitoneally and α-chloralose 50 mg/kg subcutaneously); 10 minutes later, halothane was discontinued. Following a 1-hour stabilization period, four dialysate fractions were collected into microcentrifuge tubes at 20-minute intervals; the next four samples were collected at 10-minute intervals, with the last 10-minute sample corresponding to the 10-minute infusion into the nbm. The collected dialysates were frozen immediately and kept at −80°C until high-performance liquid chromatography (HPLC) analysis could be performed for ACh.
ACh analytic procedure.
ACh in the dialysates was determined by HPLC with electrochemical detection as previously described (Beley et al., 1987). In brief, HPLC analysis was performed upon a reverse-phase column (Merck, Lichrocart 100 RP-18 encapped; 4 μm pore size; 2 × 125 mm) with a mobile phase made of 50 mM KH2PO4 (pH 7.4), 400 mg·L−1 tetramethylammonium chloride and 200 mg·L−1 heptane sulfonic acid. ACh, chromatographically separated from endogenous choline, was converted into betaine in a postcolumn reactor containing covalently bonded acetylcholine esterase and choline oxidase. Because of the action of these enzymes, ACh was first hydrolyzed into acetate and choline, and choline was subsequently oxidized into betaine and hydrogen peroxide. The amount of hydrogen peroxide, proportional to the amount of ACh released, was measured after oxidation at a potential of +0.3 V using an electrochemical detector (Coulochem II; ESA) equipped with a platinum working electrode versus a palladium reference electrode. The chromatograms were displayed on a computing integrator (Kontron, Model TIT2). ACh recovery was assessed in vitro by calibrating each probe prior to its initial use. The probe was immersed in a Ringer solution containing 500 nM ACh (Sigma, France) and perfused under the same conditions as those of the in vivo experiments. The values of extracellular ACh concentrations were calculated according to the mean estimated probe recovery (21.3%).
CBF measurement and calculations.
CBF was measured in 10 cortical regions at the end of the microinjection period as previously described (Barbelivien et al., 1995), a time corresponding to the end-point of the last dialysate. Briefly, a solution of [14C]IAP (0.111 MBq dissolved in 0.7 mL of 0.9% NaCl) was infused intravenously at a constant rate of 15 μL·s−1. During the infusion of IAP, arterial blood was sampled every 3 to 4 seconds. Approximately 35 seconds after the onset of the [14C]IAP infusion, animals were subjected to euthanasia by a bolus injection of thiopental followed by decapitation. The brains were rapidly removed, and cortical regions were dissected on ice in less than 2 minutes from the time of decapitation. Arterial samples were centrifuged, and supernatant aliquots were counted for radioactivity. Dissected brain tissues were weighted and transferred into scintillation vials; after solubilization of the tissues in 500 μL of 1 M NaOH (50°C, 12 hours), the samples were also counted for radioactivity. CBF values (mL·100 g−1·min−1) were calculated from the tissue radioactivity and the time-course of the plasma concentration of the tracer according to Sakurada et al. (1978).
Statistics
Results are expressed as mean ± SEM. The effect of the degeneration of the cholinergic basalocortical pathway from the cortex on galanin immunoreactive material was assessed through the comparison of the immunolabeling surface (μm2) in the ipsi- and contralateral nbm of sham-operated and MCA-occluded rats with a paired Student's t-test. The effect of the degeneration of the cholinergic basalocortical pathway from the cortex upon densities of each binding site type were compared through the use of repeated measures ANOVA with “group” as a between-subject factor and “side” and “structures” as within-subject factors. When significant principal factors and interactions were observed, this procedure was followed by two- or one-way ANOVA and, when necessary, by post hoc multiple comparison tests (PLSD of Fisher). Correlations between levels of each type of binding sites within the nbm were also performed. In each experimental paradigm, CBF and CGU values from the various groups of animals were compared through the use of a repeated-measures ANOVA with “group” as a between-subject factor and “side” and “structures” as within-subject factors. When statistical interactions were noticed, the previous test was followed by a two- or one-way ANOVA and, when necessary, by post hoc multiple comparison tests (PLSD of Fisher) to detect differences between groups and paired Student's t-tests to perform interhemispheric comparisons. The effect of injections into the nbm upon cortical ACh release was assessed by means of a paired Student's t-test, which compared the extracellular ACh concentration before and during the injection period. The criterion for statistical significance was taken to be P < 0.05.
RESULTS
Experiment 1: Lesion of the basalocortical system from the cortex and effect upon the galaninergic system in the nbm
Extent of the ischemic damage.
Rats that underwent permanent occlusion of the MCA were characterized by an infarct volume of 40.9 ± 5.6 mm3. The ischemic tissue was exclusively localized in the ipsilateral frontoparietal somatosensory cortex, the claustrum, and a discrete portion of the basolateral part of the striatum (Fig. 1). Cortical regions surrounding the ischemic core were the frontoparietal motor and temporal auditory cortices, whereas the retrosplenial, striate, and entorhinal cortices, as well as noncortical brain regions including the nbm, were distant from the ischemic insult. Except for a discrete necrosis localized at the level of the craniotomy (1.3 ± 0.4 mm3), no histologic damage could be observed in the sham-operated animals.
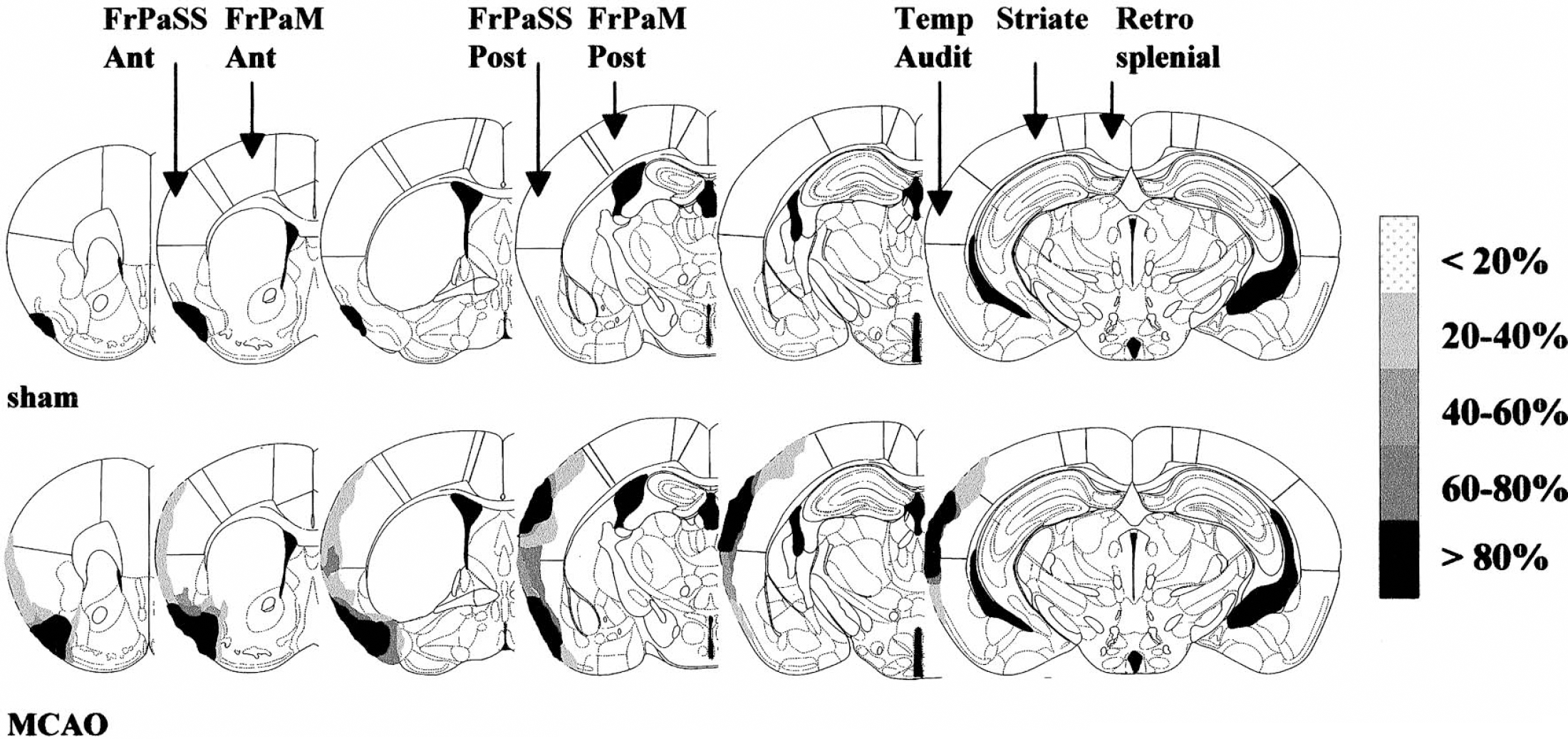
Schematic representation of the spatial distribution of the infarcted areas in sham-operated (sham) and middle cerebral artery occluded rats (MCAO) at six levels of the rat brain (from left to right: interaural +11.70, +10.60, +9.48, +7.20, +5.86, and +4.20 mm). The grey scale (expressed as the percentage of animals with a lesion in a specific cortical area/total number of animals) illustrates the distribution of the infarcted areas. FrPaSS Ant, frontoparietal somatosensory cortex, anterior part; FrPaM Ant, frontoparietal motor cortex, anterior part; FrPaSS Post, frontoparietal somatosensory cortex, posterior part; FrPaM Post, frontoparietal motor cortex, posterior part; Temp Audit, temporal auditory cortex; Striate, striate cortex; Retrosplenial, retrosplenial cortex.
Immunocytochemistry against galanin.
Specificity of the labeling was measured through extinction of the immunoreactive signal; this was achieved for galanin concentrations of 5.10−4 M (data not shown). The specific galaninergic immunoreactivity was mainly apparent in the form of neuronal fibers in the nbm, septum, thalamus, and hypothalamus, whereas few cell bodies could be also observed in the cortices. Occlusion of the MCA resulted in a significant increase (+105%) in the density of galaninergic fibers in the ipsilateral when compared with the contralateral nbm (31,300 ± 1,600 μm2 versus 15,300 ± 1,100 μm2; P < 0.0001, paired Student's t-test) while such variation could not be observed in the sham-operated rats (15,900 ± 1,200 μm2 versus 15,300 ± 1,400 μm2; P < 0.710, paired Student's t-test) (Fig. 2). Examination of the preparations allowed us to conclude that such an increase in galanin labeling was 1) irrespective of the three anteroposterior levels analyzed and 2) the result of an increase in the number of galaninergic fibers, although an increase in galanin-immunoreactivity per fiber could not be ruled out. No galanin-immunoreactive somata could be observed in the nbm of the sham and lesioned animals.
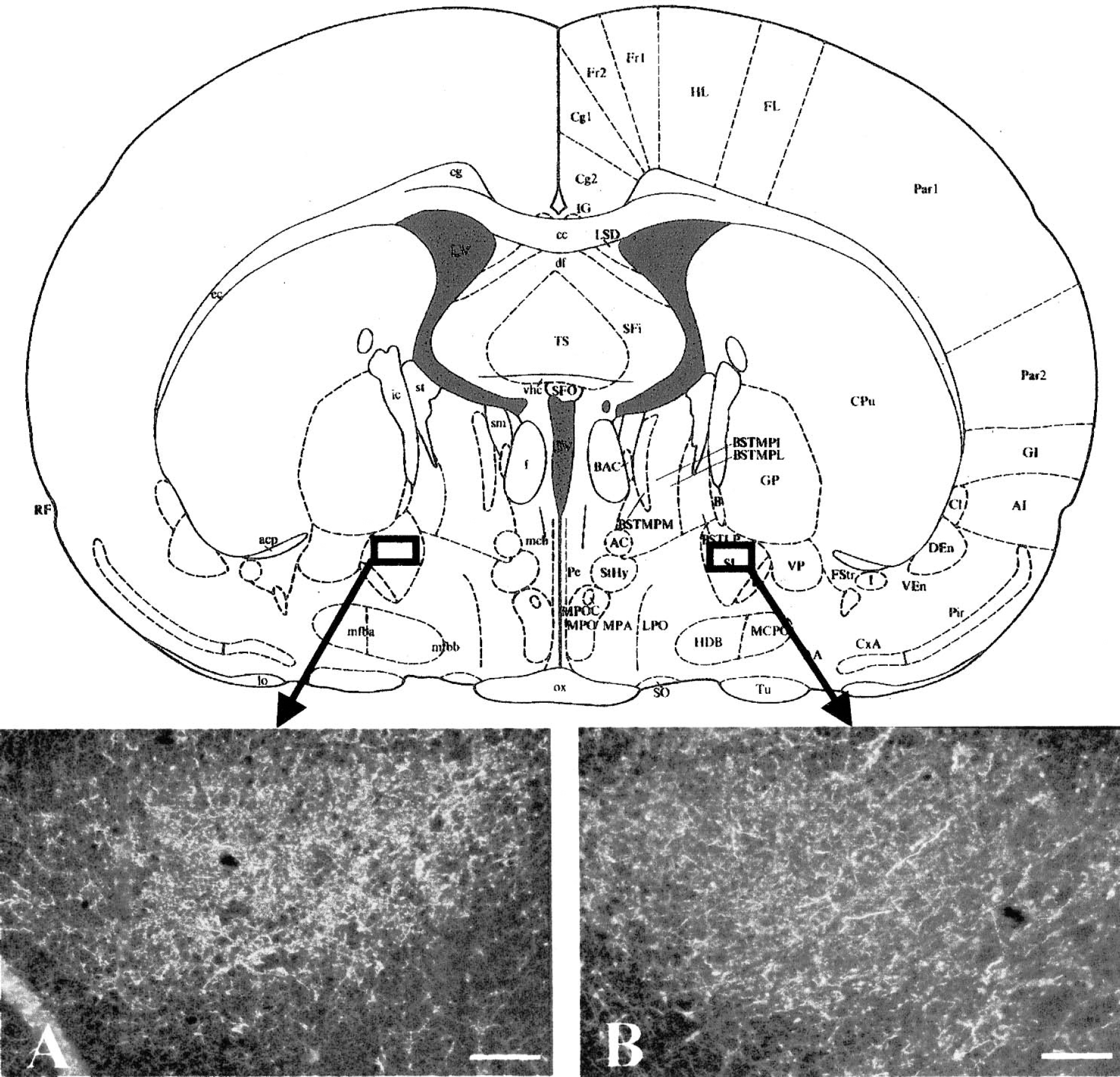
Effect of the retrograde degeneration of the basalocortical pathway upon the density of galaninergic fibers in the nucleus basalis magnocellularis of the rat. Representative example of the immunoreactive labeling against galanin in the nbm (at AP +8.20) ipsilateral to the ischemic frontoparietal cortex (A) and the nbm contralateral (B) to the ischemic frontoparietal cortex of a MCA occluded rat. Note the higher density of Gal immunoreactive labeling on the ipsilateral side. Scale bar, 50 μm. For statistical comparison, see Results.
Density of Gal-R and M1 and M2 muscarinic receptors.
Galaninergic binding sites.
Analysis of the galaninergic binding site densities revealed significant differences between structures (P < 0.0001) and a significant structure × group interaction (P < 0.01). In the ischemic core, the density of galaninergic binding sites were found to be reduced in the superficial layers of the ipsilateral frontoparietal somatosensory cortices and the claustrum (−35% and −65%, respectively; P < 0.05 ANOVA and PLSD of Fisher) (Fig. 3 and Table 1) but not in the striatum. In contrast, a significant increase (+76%, P <0.05, ANOVA and PLSD of Fisher) (Table 1) in galaninergic binding density was found in the ipsilateral nbm. No other variations could be found in the other brain regions investigated, although trends (+10–15%) were apparent ipsilaterally in the intermediate and deep layers of the frontoparietal somatosensory cortex, the septum, and the diagonal band of Broca (Table 1).
Effect of retrograde degeneration of the basalocortical pathway on the density of galaninergic, M1 and M2 muscarinic binding sites in the rat brain.
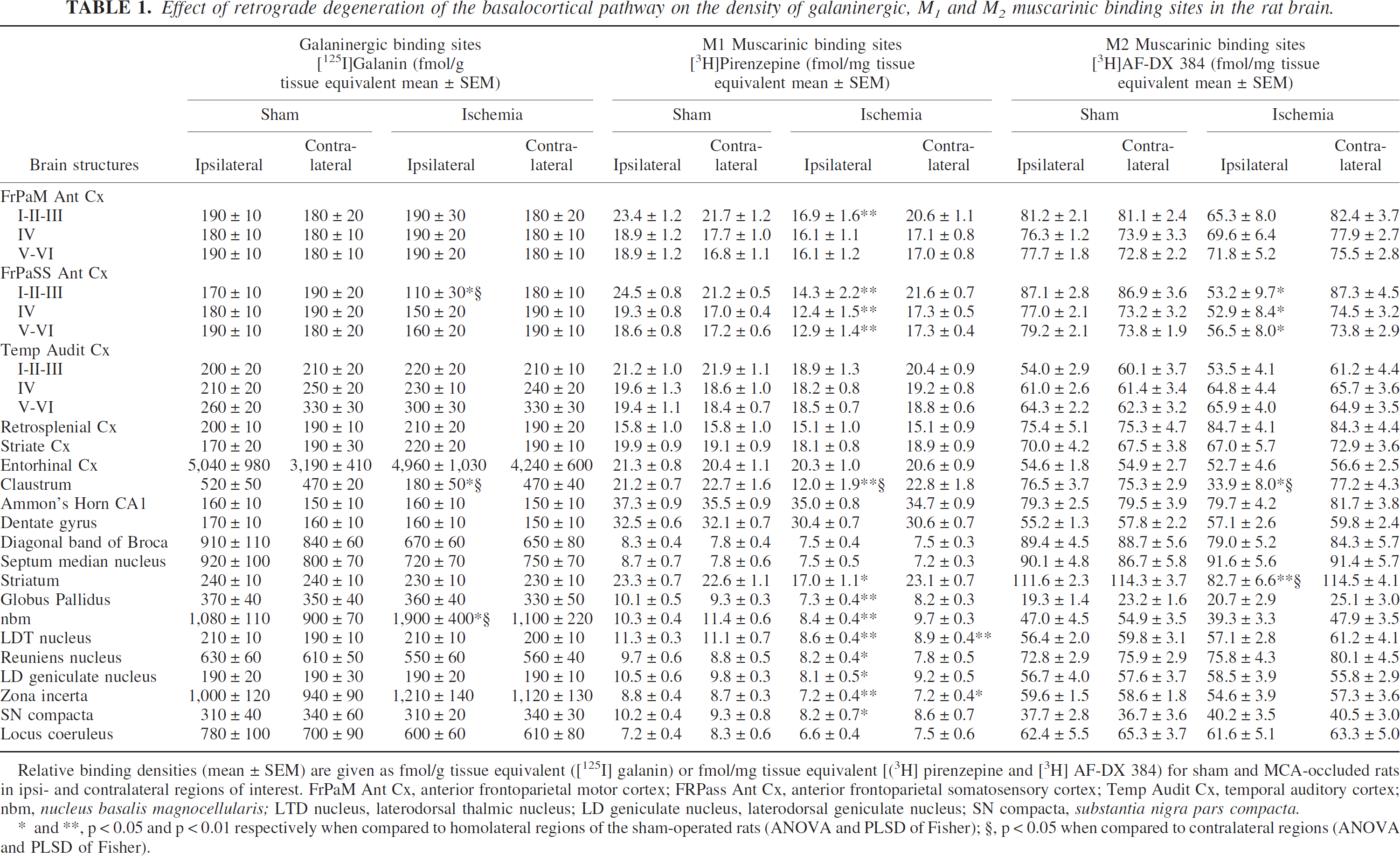
Relative binding densities (mean ± SEM) are given as fmol/g tissue equivalent ([125I] galanin) or fmol/mg tissue equivalent [(3H] pirenzepine and [3H] AF-DX 384) for sham and MCA-occluded rats in ipsi- and contralateral regions of interest. FrPaM Ant Cx, anterior frontoparietal motor cortex; FRPass Ant Cx, anterior frontoparietal somatosensory cortex; Temp Audit Cx, temporal auditory cortex; nbm, nucleus basalis magnocellularis; LTD nucleus, laterodorsal thalmic nucleus; LD geniculate nucleus, laterodorsal geniculate nucleus; SN compacta, substantia nigra pars compacta.
, p < 0.05 and p < 0.01 respectively when compared to homolateral regions of the sham-operated rats (ANOVA and PLSD of Fisher);
, p < 0.05 when compared to contralateral regions (ANOVA and PLSD of Fisher).
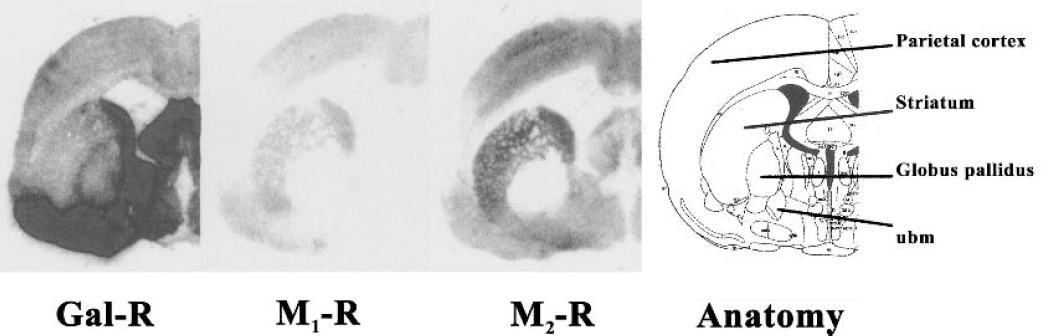
Representative illustration of the cerebral distribution of galanin (Gal-R), muscarinic M1 (M1-R), and M2 (M2-R) binding sites at one level of the brain of a MCA occluded rat, along with the respective anatomic information (AP +8.20). Autoradiograms (total binding) have been obtained on 14 μm distant coronal sections. nbm, nucleus basalis magnocellularis.
Muscarinic M1and M2 binding sites.
Analyses of the data obtained for the two cholinergic markers both revealed a principal factor (structure; P < 0.0001 and P < 0.0001 for M1 and M2 binding site densities, respectively) and a significant structure × group interaction (P < 0.001 and P < 0.01). In the ischemic core, the density of muscarinic receptor subtypes was found to be significantly reduced; one can note that both postsynaptic M1 and presynaptic M2 receptors are reduced in the frontoparietal somatosensory and motor cortices (−28 to −42%), the claustrum (−43 to −56%), and the striatum (−26 to −27%) (Fig. 3 and Table 1). In the nbm, M1 muscarinic binding sites were significantly reduced (−18%; P < 0.01, ANOVA and PLSD of Fisher) (Table 1), whereas only a nonsignificant trend was apparent for M2 receptors. A global reduction of M1 muscarinic binding sites was also apparent in the ipsilateral globus pallidus (−28%, P < 0.01, ANOVA and PLSD of Fisher), thalamic nuclei (−15 to −24%, P < 0.05 at least, ANOVA and PLSD of Fisher), and the substantia nigra pars compacta (−20%, P < 0.05, ANOVA and PLSD of Fisher); in these regions, M2 muscarinic receptors were not affected (Table 1).
Within the nbm of MCA occluded rats, a statistical negative correlation could be evidenced between M1 muscarinic binding sites (but not M2) and Gal-R densities (P < 0.01); when ipsilateral to contralateral differences were compared, the negative correlation was found to be highly significant (P < 0.001).
Experiment 2: Functional effects of galanin injections in the nbm of the rat
Measurements of CBU and CBF.
The ANOVA performed upon CBF data revealed significant differences between brain regions (P < 0.0001); based upon the presence of a significant structure × group interaction (P < 0.01), two- and one-way ANOVAs were performed to test the group effect. Injection of galanin into the nbm failed to affect CBF values when compared with sham rats except in the ipsilateral parietal motor cortex (P < 0.05) where CBF was significantly reduced when compared with shams (Table 2).
Effect of galanin injection into the nucleus basalis magnocellularis upon cerebral blood flow (ml·100 g−1·min−1) and cerebral glucose use (μmol·100 g−1·min−1) in various brain regions
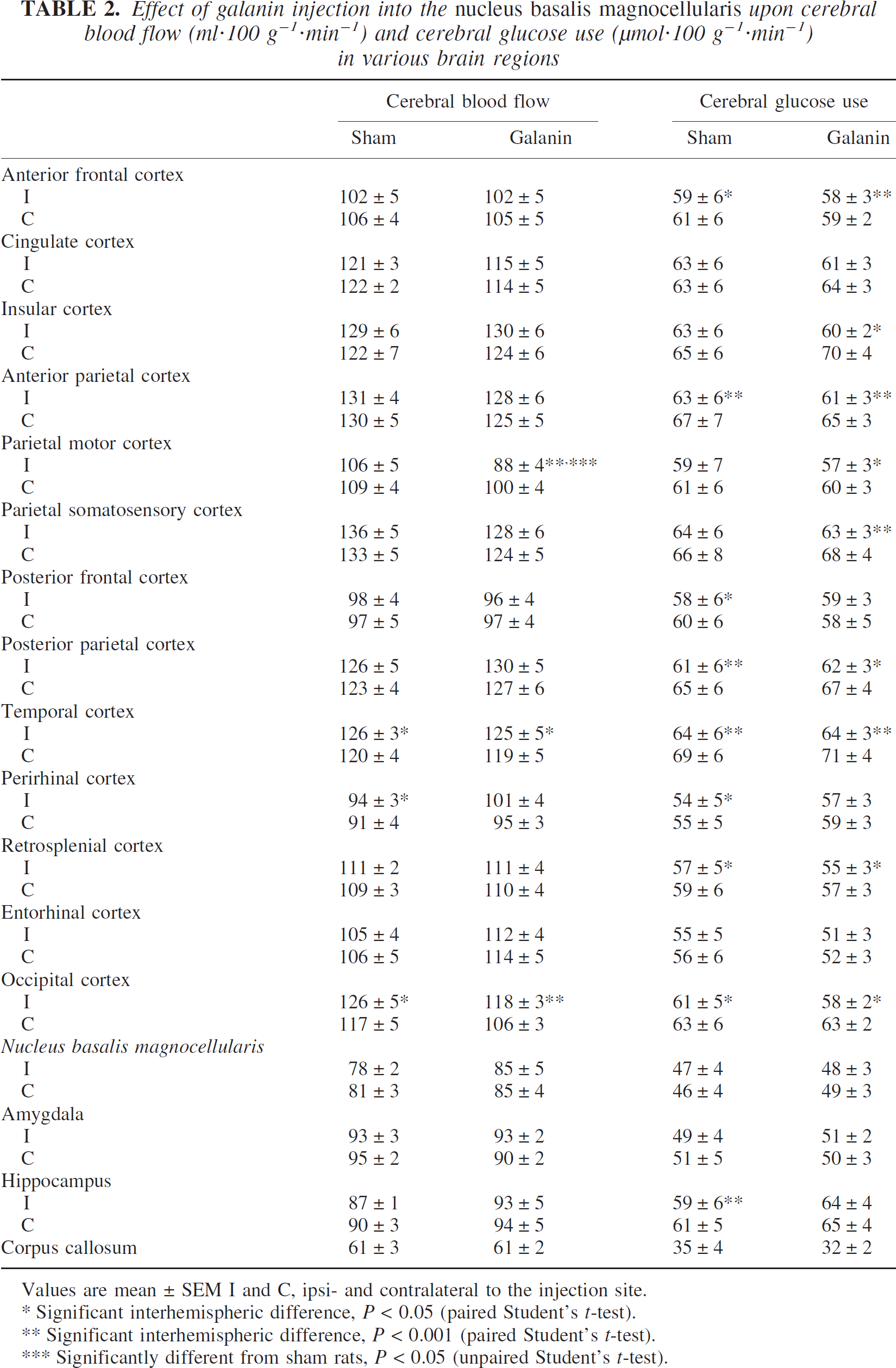
Values are mean ± SEM I and C, ipsi- and contralateral to the injection site.
Significant interhemispheric difference, P < 0.05 (paired Student's t-test).
Significant interhemispheric difference, P < 0.001 (paired Student's t-test).
Significantly different from sham rats, P < 0.05 (unpaired Student's t-test).
The ANOVA performed upon CGU values revealed significant differences between brain regions (P < 0.0001) but not between groups; because no significant structure × group interaction could be observed, no post hoc analyses could be undertaken. Thus, injection of galanin into the nbm did not affect CGU values when compared with the sham group (Table 2) in any of the brain regions investigated.
Measurements of cortical ACh release.
Microinjections of carbachol into the nbm induced a significant increase in the extracellular ACh concentration in the ipsilateral parietal cortex (+27%, P < 0.05), whereas injection of 0.9% NaCl failed to modify ACh release (Fig. 4). Coinjection of carbachol and galanin, as well as injection of galanin alone, did not modify the extracellular concentration of ACh in this cortical area (Fig. 4); in this respect, the carbachol-induced increase in cortical ACh release was totally abolished by galanin.
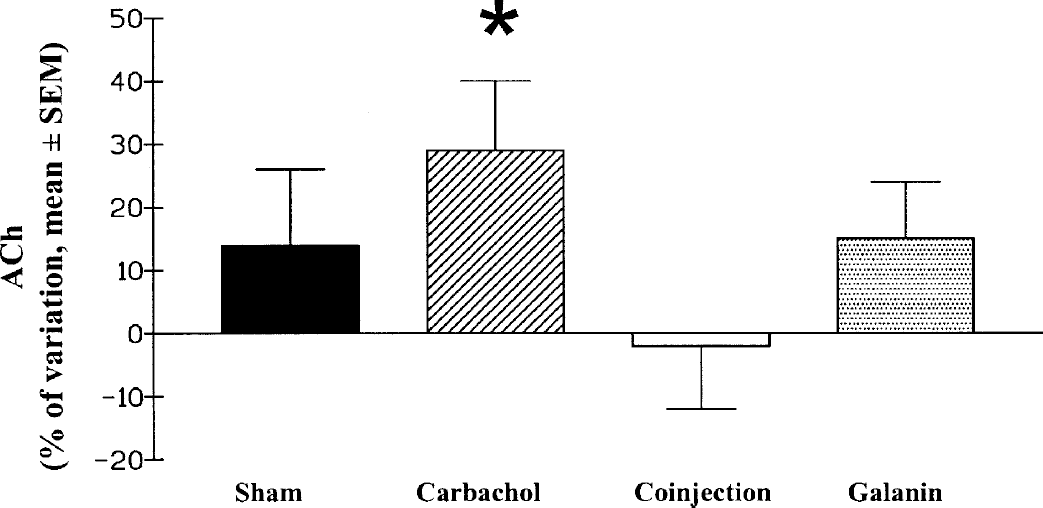
Effect of the various microinjections into the nucleus basalis magnocellularis upon acetycholine release measured in the ipsilateral parietal cortex. Values (mean ± SEM) are expressed as the percentage of the content of acetylcholine before the injection. *Significantly different from the value before microinjection (P < 0.05, paired Student's t-test).
In a parallel manner, microinjection of carbachol into the nbm induced significant increases in CBF, when compared with the three other groups, in ipsilateral cortical areas such as the anterior frontal, cingulate, temporal, and entorhinal cortices (from +18% to +33%, P < 0.05 at least) (data not shown). Coinjection of carbachol and galanin failed to modify cortical CBF when compared with the sham group. In this situation, the vasodilatatory responses noted in the ipsilateral and contra-lateral cortices after cholinergic stimulation were significantly reduced when compared with carbachol-stimulated rats. The microinjection of galanin by itself did not affect CBF in any cortical region investigated (data not shown).
DISCUSSION
Experiment 1: Lesion of the basalocortical system from the cortex induces an increase in galaninergic markers in the nbm
This study demonstrates that lesion of the basalocortical system from the cortex induces an increase in galaninergic markers in the nbm. Because previous works only measured galanin immunoreactivity or mRNA after brain lesions (Cortés et al., 1990;de Lacalle et al., 1997;Unger and Schmidt, 1993), our study is the first to our knowledge to reveal parallel, damage-induced increases in galaninergic fibers and receptors in a same region. Moreover, our binding site study suggests that the increase in galanin fiber density in the nbm could be functional. Gal-R alterations linked to the galaninergic hyperinnervation could thus participate to adaptative mechanisms and enhance the neuronal responsiveness to galanin. Because galanin expression is increased locally after lesion of the cholinergic basalocortical pathway from the cortex, one may hypothesize that galanin action is amplified by the increase in the density of galanin fibers as well as by the increase in galanin binding sites.
The relationship between the lesion and the galaninergic hyperinnervation is, however, still unclear. Indeed, electrocoagulation, but not excitotoxic lesion, of the ventral hippocampus increased the number of galanin mRNA- and immunoreactive cells in the medial septum (Cortés et al., 1990), whereas a local galaninergic up-regulation could also be found after excitotoxic lesion of the medial septum/diagonal band complex (de Lacalle et al., 1997) or the nbm (Unger and Schmidt, 1993). Galanin fibers are also increased in a structure endowing a retrograde degeneration (see further discussion). Altogether, these results suggest that the increase in galaninergic markers in the nbm can be triggered both by axonal damage or direct and indirect damage to somas.
Whether the galaninergic upregulation in the basal forebrain is associated to the damage of local cholinergic neurons has been hypothesized (Hartonian et al., 2002). Indeed, cortical infarction or devascularization in rats and primates leads to cholinergic alterations in the nbm/NBM (Figueiredo et al., 1993;Kataoka et al., 1991;Liberini et al., 1994). One might argue that such local damage in the nbm could be the result of a retrograde degeneration of ascending basalocortical fibers or an anterograde degeneration of descending corticobasal neuronal processes. Although an anterograde mechanism cannot be ruled out, a retrograde mechanism appears to be involved, at least partly, on the basis of the literature (Figueiredo et al., 1993;Kataoka et al., 1991;Liberini et al., 1994). Such is also strongly supported by our present demonstration of a reduction, at the level of the infracted and surrounding cortices, of the binding densities of M1 and M2 muscarinic receptors, respectively known to be localized on postsynaptic and presynaptic elements of the basalocortical pathway (Vilaro et al., 1994). One might further propose that the retrograde degeneration mainly alters cholinergic basalocortical neurons rather than other ascending fibers such as GABAergic and glutamatergic neurons, because these latter mainly project to the enthorinal cortex (Manns et al., 2001) where no cellular or neurochemical alteration could be found in the present study. At the level of the nbm, the literature is, however, unclear as to the putative preferential localization of M1 and M2 muscarinic receptors on either local cells and dendrites or presynaptic terminals coming from outside the nbm (Decossas et al., 2003;Levey et al., 1995;Mufson et al., 1998;Vilaro et al., 1994;Zilles et al., 1991). It should thus be interesting to support such a hypothesis through the direct quantitation of cholinergic cell bodies by either ChAT or vesicular acetylcholine transporter immunoreactivity. Nevertheless, our results, in association with the local increase in galanin immunoreactivity and the strong correlation between the local decrease in M1 receptors and the increase in Gal-R, are in favor of a direct link between cortical infarction, damage, or loss of the nbm cholinergic neurons and the local increase in galaninergic markers.
Our results interestingly parallel the situation observed in the NBM of patients with AD, in which galanin or Gal-R are increased (Bowser et al., 1997;Chan-Palay, 1988;Mufson et al., 2000) but without reduction in m2-immunoreactive neurons (Mufson et al., 1998). This could thus suggest that the galaninergic hyperinnervation in the NBM of patients with AD may result partly from the retrograde degeneration of the basalocortical cholinergic system. Several hypotheses have been proposed to explain the damage and degeneration of the NBM cholinergic neurons in AD (Henderson, 1996); indeed, cell shrinkage, downregulation of ChAT (Perry et al., 1982), and retrograde degeneration mechanisms such as those illustrated by the reduction in neurotrophin receptors and retrograde NGF transport by NBM cholinergic neurons (Mufson et al., 1995) might thus contribute to the degeneration of basal forebrain neurons in AD.
Experiment 2: Functional consequences of the increase in galaninergic markers in the nbm
To study the putative functional consequences of such an increase in galaninergic innervation in the nbm, one might wait for the measurements of cortical activity indices in the rats that exhibit this galaninergic pattern in the nbm, that is, in the MCA occluded rats. However, the ischemic model used in the first part of this study is linked to direct necrosis of the frontoparietal cortex that in turn directly affects local activity. We thus quantified cortical activity indices such as ACh release, CBF, and CGU (Barbelivien et al., 1999) in response to galanin infusion in the nbm, hypothesized to mimic a local increase in galanin transmission, either at rest or under cholinergic coactivation. With such a paradigm, galanin infused alone into the nbm failed to affect cortical ACh release, CBF, or CGU at rest but abolished the increases in cortical ACh release and CBF elicited by the parallel activation of the cholinergic basalocortical system. Altogether, this demonstrates that galanin exhibits its effect(s) only when the cholinergic basalocortical system is activated.
Such a pattern should be of importance in pathophysiologic conditions. Indeed, hypometabolism and hypoperfusion have been demonstrated under resting conditions in AD (Friedland et al., 1983;Prohovnik et al., 1988). Moreover, vasomotor responses to cognitive tasks, known to be dependent upon the basalocortical cholinergic pathway (Pardo et al., 1991;Prohovnik et al., 1997;Sarter and Bruno, 1997), are also altered in AD (Meguro et al., 2001). In this respect, galanin and its increased innervation in the nbm do not appear to be of importance in the resting reduction of CBF and CGU observed in AD, based upon the fact that this peptide had neither a direct effect upon nbm neuronal activity nor an indirect effect upon the local metabolic activity that, in turn, would affect neuronal activity indices in the areas of projections (Barbelivien et al., 1995, 1998, 1999). One cannot rule out, however, the fact that galanin may interact with nbm cholinergic neurons only when these latter are altered and putatively hyperactive. Such a situation should exist in AD, in which, in parallel to the galaninergic hyperinnervation of the nbm (Bowser et al., 1997;Chan-Palay, 1988), one might hypothesize an increase in galanin release based upon 1) the coexistence of galanin and ACh in some neurons of the human NBM (Chan-Palay, 1988) and 2) the increase in firing rate of the surviving cholinergic neurons, thought to compensate for their degeneration, as proposed by Consolo et al. (1994). On this basis, the increase in galanin and galanin receptors should exacerbate the inhibition of the remaining cholinergic neurons and limit the activation-elicited increases in cortical activity indices in patients with AD (Meguro et al., 2001;Pardo et al., 1991;Prohovnik et al., 1997) because cortical ACh release could be reduced both by an action of galanin within the nbm (present results) and in the cortex (Wang et al., 1999). In other words, functioning of the remaining cholinergic NBM neurons in AD might be thus inhibited by the increase in galaninergic markers in the NBM and the increased release of galanin in the cortex. Such a conclusion could find support from the demonstration that basal forebrain lesion-induced cognitive impairments are reduced by coadministration of a M1 muscarinic agonist and a Gal-R antagonist (McDonald et al., 1998). This latter result is in line with a putative deleterious role for the galaninergic hyperinnervation in AD. Galanin has, however, also been presented to exert beneficial effects when upregulated after neuronal damage or in neurodegenerative/regenerative processes (Cortés et al., 1990;de Lacalle et al., 1997;Holmes et al., 2000). Galanin, moreover, plays a survival role in the development of the forebrain cholinergic system (O’Meara et al., 2000). Additionally, the fact that NGF induces an up-regulation of galanin in the basal forebrain (Planas et al., 1997) might be in favor for the hypothesis of a beneficial role of galanin in regenerative processes. Overall, the increase in expression of galanin could thus be deleterious for the functioning of the nbm cholinergic system but salutary for the survival of these neurons.
CONCLUSION
The present study suggests that the increased galaninergic innervation induced within the nbm/NBM by the degeneration of the basalocortical pathway from the cortex might result in an exacerbation of the cholinergic deficits found in neuropathologies such as AD. Additional work is, however, needed to refine the model of hyperinnervation of galaninergic fibers and receptors. Indeed, although the ischemic insult undertaken in our study was mild, such a model might appear quite far from AD where a putative discrete degeneration of cortical neuronal terminals or somata could be characteristic. One might be also interested in the chronic demonstration of an increase in galaninergic expression in aged animals because neurologic changes seen in AD are slow, progressive, and linked to aging.
Footnotes
Acknowledgment
The authors thank Mr. D. Divoux and Dr. L. Besret for technical assistance, Pr. G. Tramu for the gift of anti-GAL antibody, and the cyclotron and radiochemistry staff of Cyceron for the syntheses of [18F]FDG.