
Abstract
The single-cell gel electrophoresis (comet assay) was used to evaluate the possibility of detecting single-strand breaks of brain DNA in the early phase of ischemia. Four hours after occlusion of the middle cerebral artery (MCAO) in rats, the percentage of DNA migrating into the comet tail (indicating the presence of breaks) increased from 11.4 ± 4.70 to 34.7 ± 9.2 (means ± SD) in the caudate and from 9.9 ± 4.3 to 42.8 ± 14.1 in the cortex. Interestingly, a subpopulation of cells exhibiting higher resistance to the ischemic insult was present in the caudate putamen, but not in the cortex. Administration of MK801, an N-methyl-
The single-cell gel electrophoresis, or the “comet” assay, is a sensitive method initially developed as a modification of the alkaline elution technique for the study of DNA breakage (Singh et al., 1988). It involves embedding individual cells in agarose on a microscope slide and measuring the degree of migration of nuclear DNA on electrophoresis. The extent of migration is proportional to the number of breaks in DNA, and its evaluation allows indirect measurement of the number of DNA breaks at the single-cell level. This procedure has been widely used for genotoxicity studies (McKelvey-Martin et al., 1993) and for monitoring the exposure to DNA-damaging agents in human populations (Moller et al., 2000). It has also been applied to neurons exposed to genotoxic compounds or electromagnetic fields (Malyapa et al., 1998; Svedenstal et al., 1999; Tsuda et al., 2000), but was never used to investigate DNA breaks in postischemic brain damage or other neurologic disorders.
In animal models of postischemic neuronal damage, single-strand breaks (SSBs) and oxidized bases in brain DNA are rapidly formed because of excessive formation of free-radical species (Liu et al., 1996; Chen et al., 1997; Love, 1999; Nagayama et al., 2000). A central role in the initiation of the cascade of molecular events leading to the formation of these reactive species and to neuronal damage in brain ischemia is played by glutamate, which is massively released in the extracellular space by the ischemic insult (Benveniste et al., 1984; Simon et al., 1984; Lombardi and Moroni, 1992; Szatkowski and Attwell, 1994). The increase in intracellular Ca2+ concentration triggered by the activation of glutamate receptors causes in turn the activation of intracellular Ca2+-dependent signaling cascades that contribute to further free radical formation, energy depletion, and eventually lead to neuronal cell death (Choi, 1988a; Pellegrini-Giampietro et al., 1990; Dawson et al., 1991). Single-strand breaks have been shown to activate the DNA repair signaling protein poly(ADP-ribose) polymerase (PARP) (Zhang et al., 1994; de Murcia and Menisser-de Murcia, 1994; Eliasson et al., 1997), whose overactivation with subsequent cellular energy and NAD+ depletion is thought to make a major contribution to the progression toward irreversible neuronal damage in animal models of cerebral ischemia (Endres et al., 1997; Eliasson et al., 1997).
In the later stages of brain ischemia, when cellular death becomes prominent, a second type of DNA damage arises from the action of endogenous Ca2+-activated endonucleases, which bring about DNA fragmentation (Charriaut-Marlangue et al., 1995). At this stage, double-strand breaks (DSBs) are detected in the ischemic regions (Chen et al., 1997).
The aim of this study was to evaluate the possibility of using the comet assay for the study of the early, oxidative stress-induced DNA damage in cerebral ischemia. In fact, given the sensitivity of the method, the comet assay appears to be suitable for the early detection of low levels of damage. We conducted this investigation in a widely used rat model of cerebral focal ischemia, that is, the monolateral occlusion of the middle cerebral artery, and measured DNA breaks after 2 and 4 hours of ischemia in the territories perfused by the middle cerebral artery, the parietal cortex, and the caudate putamen. The same areas in the contralateral side and the hippocampus, a region outside the middle cerebral artery territory, served as control for the basal levels of DNA breaks.
MATERIALS AND METHODS
All of the experiments were approved by an ethics committee of the University of Florence, and animal housing and handling followed the guidelines of the European Community's Council for Animal Experiments. Male Sprague Dawley rats (body weight 280 to 300 g) purchased from Morini (Italy) were used.
Middle cerebral artery occlusion
Middle cerebral artery occlusion (MCAO) was induced by using a previously described, relatively noninvasive technique (Longa et al., 1989). Anesthesia was induced with 2% isoflurane in air and maintained with the lowest active concentration of anesthetic. Body temperature was measured with a rectal probe and kept at 37°C with a heating pad. The external and internal right carotid arteries were dissected under an operating microscope, and a silk suture was tied loosely around the external carotid stump. A silicone-coated nylon filament (diameter: 0.28 mm) was then inserted through the external into the internal carotid artery up to the Willis circle to occlude the right middle cerebral artery. The silk suture was finally tightened around the intraluminal filament to prevent bleeding. To maintain the animals at standard temperature conditions, a heating pad was used for the entire duration of the experiments (2 or 4 hours).
For the measurement of the infarct areas and volumes, the animals had 2 hours of MCAO, and 48 hours after surgery they were injected with chloral hydrate (400 mg/kg) and transcardially perfused with 5 mL of saline containing 2% 2,3,5-triphenyltetrazolium chloride (Sigma, Milan, Italy). Brains were removed 20 minutes later and fixed in 4% paraformaldehyde. One-millimeter-thick coronal sections were prepared and the infarct areas were measured using a computerized image analysis system (Image-Pro Plus 3.0, Silver Spring, MD, U.S.A.). The infarct volume was calculated as described (Cozzi et al., 1999; Li et al., 2000).
Administration of MK801 or DPQ
The N-methyl-
Tissue dissection and nuclei preparation
The animals were killed by decapitation at different times after MCAO, and the brains were rapidly transferred into ice-cold, oxygenated (100% O2) Hanks' balanced salt solution [composition in mmol/L: NaCl 137, KCl 5.4, MgSO4 0.8, CaCl 1.2, NaHCO3 4.2, Na2HPO4 0.33, K2HPO4 0.4, glucose 5.5, N-(2-hydroxyethyl)piperazine-N'-2-ethanesulfonic acid 10, pH 7.4]. The parietal cortex, the hippocampus, and the caudate putamen were dissected out, weighed, and homogenized in 20 vol (wt/vol) of STM buffer [composition in mmol/L: sucrose 250, Tris-HCl 50, pH 7.4, MgSO4 5, phenylmethylsulfonyl fluoride 0.5], to obtain isolated intact nuclei (Kaufmann and Shaper, 1984).
Comet assay
Aliquots (20 μL) of the brain homogenate containing the isolated nuclei were mixed with 85 μL of melted low-melting-point agarose (Fisher Scientific, Leics, U.K.), layered on agarose-precoated microscope slides and allowed to solidify on ice for 10 minutes. Two slides were prepared for each experimental point. A further layer of low-melting-point agarose was added on top of the nuclei-containing layer, and after solidification the slides were run through the comet assay as described (Giovannelli et al., 2000). Briefly, the slides with the agarose-embedded nuclei were subjected to a lysis step (1-hour incubation at 4°C in 1% N-lauryl-sarcosine, 2.5 mol/L NaCl, 100 mmol/L Na2EDTA, 1% TritonX-100, 10% dimethylsulfoxide) to eliminate nuclear membranes, proteins, and all the non-nuclear components of the homogenate. After completion of the lysis step, the slides were placed for 20 minutes in an ice-cold electrophoresis chamber (model GNA-200, Pharmacia, Milan, Italy) containing alkaline electrophoresis buffer (300 mmol/L NaOH, 1 mmol/L Na2EDTA) to allow DNA unwinding. The electrophoresis was subsequently conducted for 20 minutes at 25 V and 300 mA. At the end of the electrophoresis, the slides were washed with neutralization buffer (40 mmol/L tris-HCl, pH 7.4), stained with ethidium bromide overnight, and analyzed on the following day.
To distinguish SSBs and DSBs, a neutral electrophoresis (Klaude et al., 1996) was occasionally performed on a separate set of slides, identical to those undergoing alkaline electrophoresis. These slides were run through the procedure along with the others until the completion of the 20-minute incubation in alkaline electrophoresis buffer. This latter step was designed to carry out complete degradation of RNA. They were then shifted to a neutral buffer (TAE: 40 mmol/L Tris-acetate, 1 mmol/L EDTA, pH 8.0), incubated for 30 minutes to allow reannealing of the DNA strands, and subjected to electrophoresis in TAE buffer (25 V, 30 mA, 20 minutes).
Microscopic analysis was carried out by means of a Labophot-2 microscope (Nikon, Tokyo, Japan) provided with epi-fluorescence and equipped with a rhodamine filter (excitation wavelength 546 nm; barrier 580 nm). The images of 50 randomly chosen nuclei per slide were captured and analyzed using a custom-made imaging software coupled with a CCD camera (model C5985, Hamamatsu, Sunayama-Cho, Japan). The amount of damaged DNA (SSBs and DSBs) migrated in the tail was expressed as percent of total fluorescence for each nucleus (percent DNA in tail). This value was then averaged over the 100 nuclei measured in the duplicate slides, and the mean ± SD of percent DNA in the tail was calculated within experimental groups. To calculate the distribution of DNA damage within each sample, we also classified the nuclei into 5 categories with increasing tail migration (class I: 0–6%; class II: 6.1–17%; class III: 17.1–35%; class IV: 35.1–60%; class V: 60.1–100%), as proposed (Wollowski et al., 1999). The percent frequency distribution into the five categories was then calculated for each duplicate and averaged within experimental groups.
Statistical analysis
Differences between groups were analyzed by means of analysis of variance and Student's t-test. Values were expressed as means ± SD.
RESULTS
Evaluation and characterization of early MCAO-induced DNA damage
In preliminary experiments, we noticed that MCAO caused, in a few hours, significant DNA damage in the rat cortex and caudate-putamen. Figure 1 shows examples of different DNA migration profiles in cells obtained from the parietal cortex of a rat with MCAO for 4 hours. The five panels show representative images of single nuclei with increasing degrees of DNA migration into the comet tail, fitting in the I to V categories described in Materials and Methods, I being undamaged and V the most damaged class.
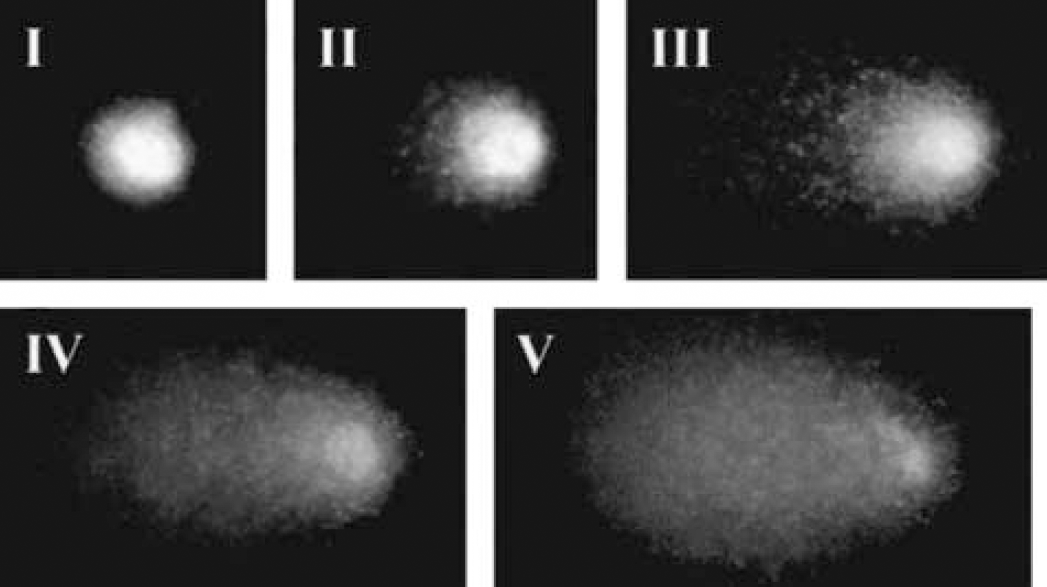
Images of ethidium bromide-stained nuclei obtained from postischemic rat cortex (4 hours middle cerebral artery occlusion), exhibiting different degrees of DNA damage after electrophoretic migration. Each image represents one class of damage as indicated in each panel. DNA damage level increases from class I to V, as indicated by the increasing tail migration (class I: 0–6%; class II: 6.1–17%; class III: 17.1–35%; class IV: 35.1–60%; class V: 60.1–100%).
To characterize the type of DNA strand breaks (single or double) found 4 hours after MCAO, samples of the parietal cortex or of the caudate putamen were run in parallel in standard alkaline and neutral buffers. Virtually no electrophoretic migration was found in neutral buffer (DNA in the tail was not different from the respective nonischemic controls), indicating that the DNA migration seen in alkaline conditions was almost exclusively caused by SSBs (Fig. 2).
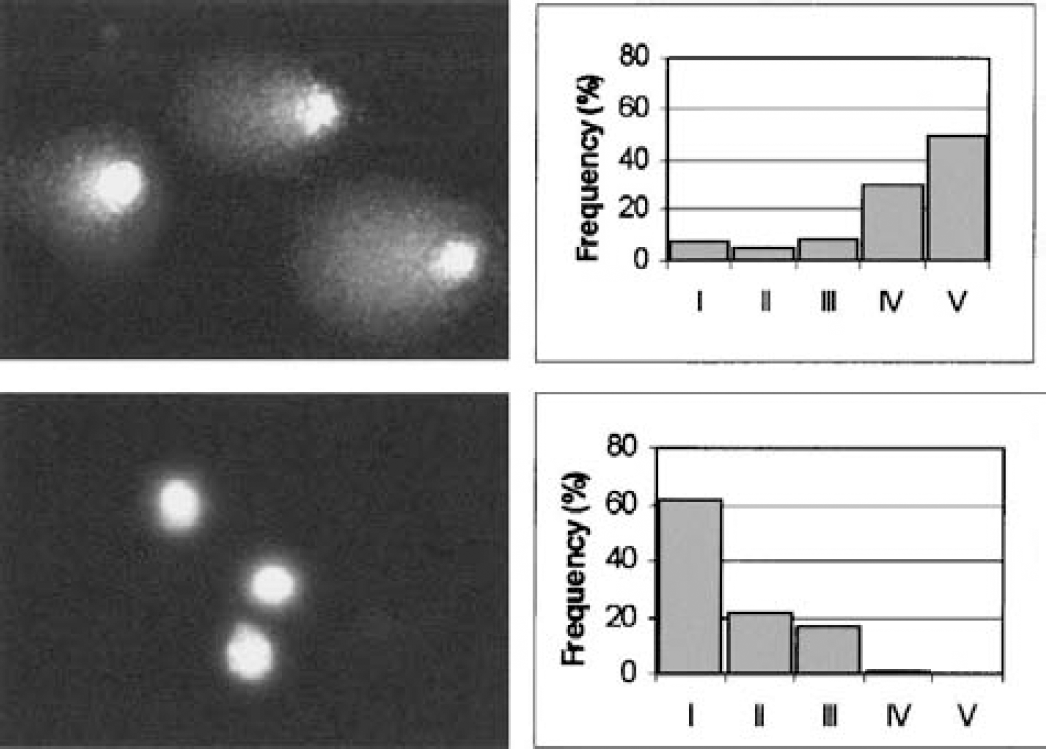
Comparison of alkaline and neutral electrophoresis to separately detect single- and double-strand DNA breaks. A sample of cortex obtained 4 hours after middle cerebral artery occlusion was run in parallel in standard alkaline (upper panels) and neutral conditions (lower panels). The images of representative nuclei after electrophoresis are shown on the left. In alkaline conditions, most of the cells exhibited a broken DNA capable of migrating in the electric field, whereas in neutral conditions, no migration was observed. The quantified data are shown in the panels on the right, where the frequency distribution into the five classes of increasing DNA damage (from class I to V) is shown for each sample.
Effects of MCAO on DNA damage
Table 1 shows the effects of MCAO on DNA damage in the parietal cortex, caudate putamen, and hippocampus. The damage was expressed as percent DNA migration in the tail, and 100 nuclei were counted in each animal for each brain region. In the caudate putamen, the damage reached a statistically significant level in 2 hours. In the parietal cortex, the percent of DNA in the tail increased 4.3-fold in 4 hours. No damage was detected in the hippocampus, a brain region outside the MCA territory. Furthermore, MCAO did not affect DNA damage in the contralateral nonischemic side, where the levels of DNA breakage were not different from those measured in the corresponding brain areas of naive animals.
Time course of the development of DNA damage in the cortex, caudate putamen and hippocampus after middle cerebral artery occlusion
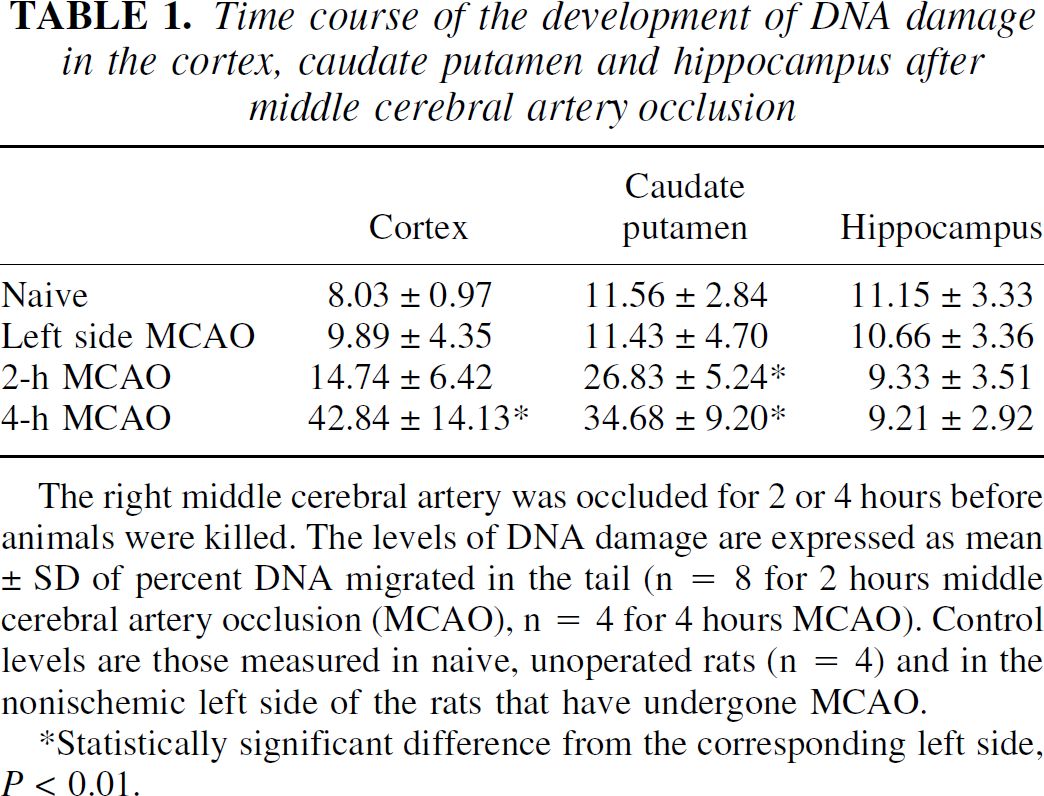
The right middle cerebral artery was occluded for 2 or 4 hours before animals were killed. The levels of DNA damage are expressed as mean ± SD of percent DNA migrated in the tail (n = 8 for 2 hours middle cerebral artery occlusion (MCAO), n = 4 for 4 hours MCAO). Control levels are those measured in naive, unoperated rats (n = 4) and in the nonischemic left side of the rats that have undergone MCAO.
Statistically significant difference from the corresponding left side, P < 0.01.
The pattern of distribution of ischemia-induced DNA damage is reported in Figs. 3 and 4. In the nonischemic side of the brain, most of the nuclei (approximately 80% of total) were undamaged or slightly damaged, as indicated by their clustering in classes I and II. This pattern was not different from that found in the corresponding brain areas of naive, unoperated animals, where the frequency distribution of the cells within the 5 categories of damage from I to V was the following: 49%, 37%, 11%, 2%, and 1% for the cortex, 38%, 43%, 13%, 5%, and 1% for the caudate putamen, and 43%, 36%, 15%, 3%, and 3% for the hippocampus.
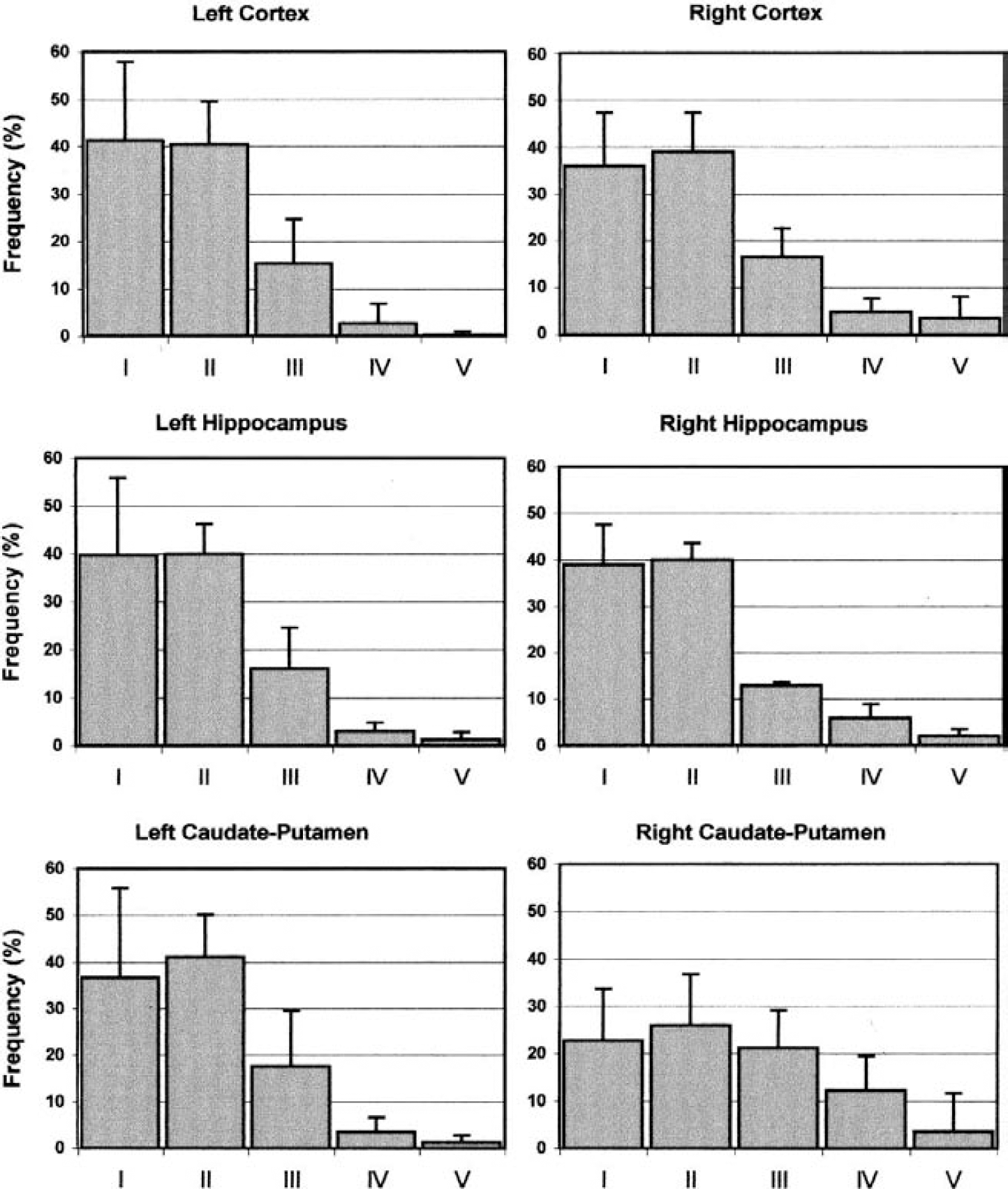
Frequency distribution of DN damage in the cortex, caudate putamen and hippocampus after 2 hours o middle cerebral artery occlusion. The percent frequency (mean ± SD) of nuclei in each class of damage (fro class I to V) is shown for each brain area in the left nonischemic side (le panels) and in the right ischemic side (right panels). n = 8 for each experimental group.
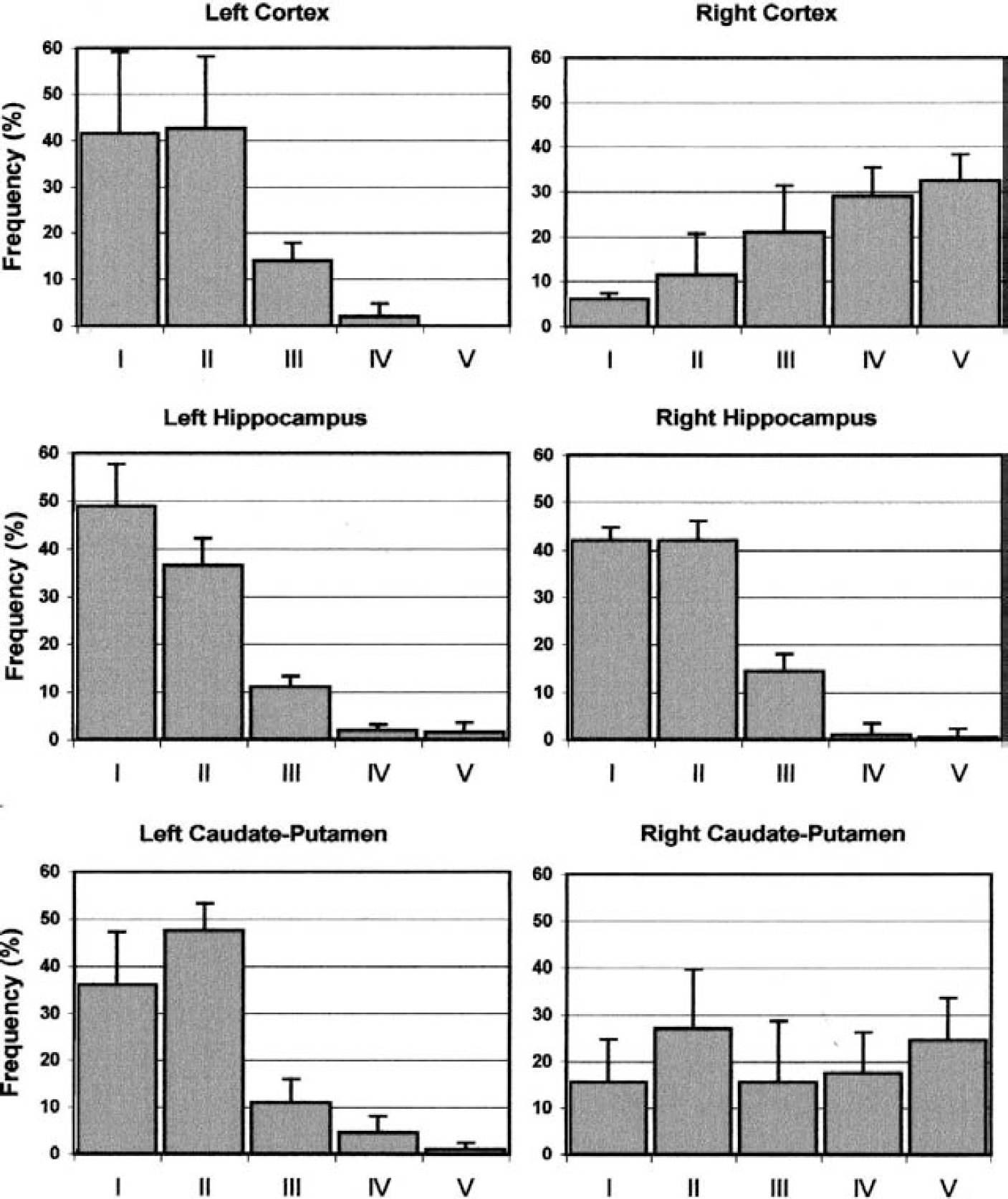
Frequency distribution of DNA damage in the cortex, caudate putamen, and hippocampus after 4 hours of middle cerebral artery occlusion. The percent frequency (mean ± SD) of nuclei in each class of damage (from class I to V) is shown for each brain area in the left nonischemic side (left panels) and in the right ischemic side (right panels). n = 4 for each experimental group.
Two hours after MCAO, it was possible to detect a minor increase in the percentage of highly damaged nuclei (class IV and V) in the cortex. This increase was particularly evident in the caudate putamen, where only approximately 45% of the total nuclei were left in the low-damage classes. Four hours after MCAO, most of the nuclei in the cortex were highly damaged, with only approximately 15% of them remaining in the low-damage classes. Also in the caudate putamen, the extent of damage was robust and more than at 2 hours (the percentage of nuclei in the high-damage classes increased from 15% at 2 hours to 40% at 4 hours). However, in this brain region, a substantial number of nuclei were still undamaged after 4 hours of ischemia (approximately 40% of total nuclei remained in classes I and II).
Effects of neuroprotective agents on postischemic DNA damage
To study whether and how putative neuroprotective agents reduced postischemic DNA damage, we tested the NMDA receptor antagonist MK-801 and the PARP inhibitor DPQ. The doses and administration schedules of these agents were selected in previous experiments to obtain a comparable degree of reduction of the infarct volumes (Fig. 5).
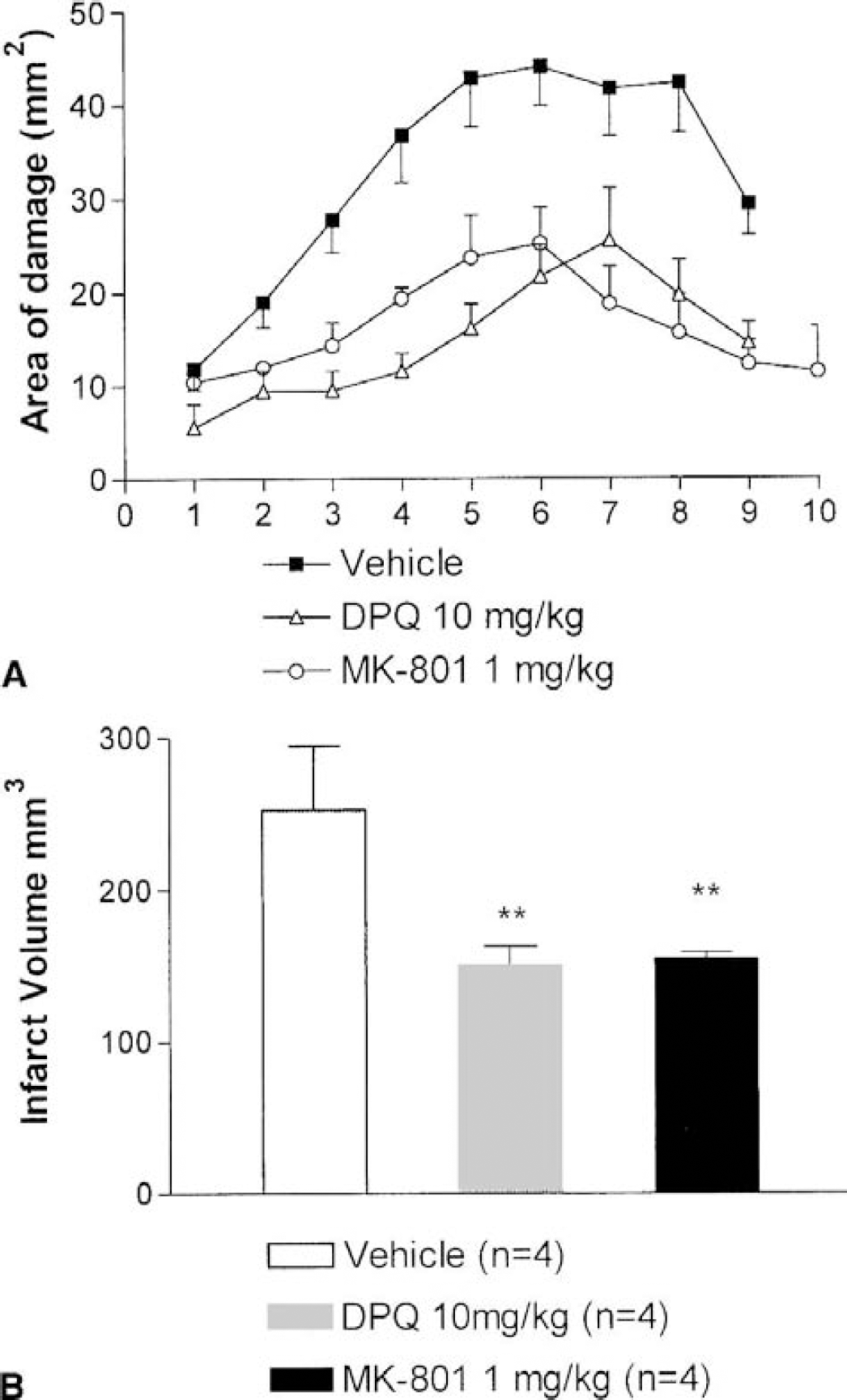
Neuroprotective effect of MK-801 (1 mg/kg subcutaneously, 10 minutes before middle cerebral artery occlusion [MCAO]) and of 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (10 mg/kg intraperitoneally, 2 hours before and 1 hour after MCAO) in rats that underwent transient (2-hour) MCAO and were killed 48 hours after the ischemic challenge. The infarct area in square millimeters at 10 different coronal levels (
Table 2 shows that the ischemia-induced DNA damage in the caudate putamen was not present in rats treated with MK-801, thus suggesting that the neuroprotective action of NMDA antagonists is associated with a reduction in postischemic DNA damage. Conversely, a neuroprotective treatment with the PARP inhibitor DPQ did not change the MCAO-induced DNA damage in the caudate putamen.
Effect of MK-801 and of DPQ on ischemia-induced DNA damage in the parietal cortex and caudate putamen after 2 hours of middle cerebral artery occlusion
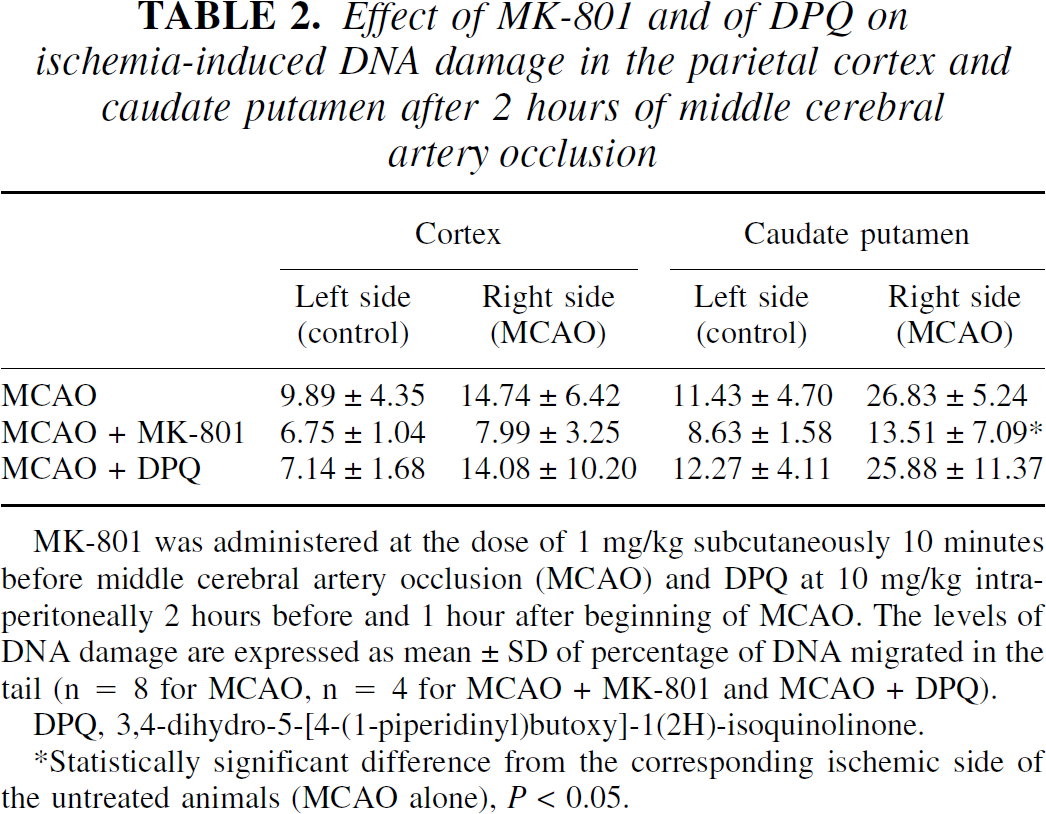
MK-801 was administered at the dose of 1 mg/kg subcutaneously 10 minutes before middle cerebral artery occlusion (MCAO) and DPQ at 10 mg/kg intraperitoncally 2 hours before and 1 hour after beginning of MCAO. The levels of DNA damage are expressed as mean ± SD of percentage of DNA migrated in the tail (n = 8 for MCAO, n = 4 for MCAO + MK-801 and MCAO + DPQ).
DPQ, 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone.
Statistically significant difference from the corresponding ischemic side of the untreated animals (MCAO alone), P < 0.05.
DISCUSSION
Our results show that the comet assay may be considered a novel and useful approach for measuring early DNA damage in cerebral ischemia. We noticed that after MCAO, single DNA breaks first appeared in the caudate putamen (where their number significantly increased after 2 hours of ischemia) and then in the cortex, where statistically significant damage was detected at 4 hours. This temporal progression of tissue injury from the caudate to the cortex is in agreement with previous reports showing that cell degeneration and death after MCAO appears first in the basal nuclei and then spreads to the cortex (Garcia et al., 1995). The levels of DNA damage detected with the comet assay were relatively low. It has indeed been proposed that it is possible to calculate the number of DNA breaks from the percentage of DNA migrated in the comet tail, referring to a calibration curve obtained in cultured cells irradiated with different X-ray doses and assuming that 1 Gy induces 0.31 breaks/109 daltons (Collins et al., 1996). On this basis, we calculated that the number of DNA breaks was 1.06 breaks/106 base pairs in the cortex and 1.25 breaks/106 base pairs in the basal nuclei under basal conditions. After MCAO, these values increased to 3.21 and 4.20 breaks/106 base pairs in the caudate putamen, and to 1.67 and 5.24 breaks/106 base pairs in the cortex, at 2 and 4 hours, respectively.
The alkaline electrophoresis that we routinely use allows detection of both DSBs and SSBs, whereas in neutral conditions only DSBs can be detected. As shown by the neutral electrophoresis experiments, in the early ischemic phases, DNA damage consisted almost exclusively of SSBs, in line with the suggestion that oxidative stress plays a key role in postischemic neuronal death.
By analyzing the distribution of ischemia-induced DNA damage within each cell group, we showed the presence, in the caudate putamen, of a subgroup of cells exhibiting significant resistance to the ischemic insult. The comet assay does not allow the identification of the cell population resistant to the ischemic insult, but the results obtained with this approach are in line with literature reports indicating significant differences in the sensitivity of striatal neurons to ischemic insults. In fact, the large aspiny cholinergic interneurons of the striatum have been shown to be resistant to oxygen and glucose deprivation (Johnston and Hudson, 1987; Burke and Karanas, 1989), possibly because of differences in the ionic mechanisms regulating membrane potentials (Centonze et al., 2001). It has also been reported that striatal NADPH-diaphorase-positive neurons are relatively resistant to excitotoxic damage and to neonatal hypoxia-ischemia (Ferriero et al., 1988).
Ischemia-induced SSBs in brain have previously been reported using in situ nick translation (DNA polymerase I-mediated biotin-dATP nick translation [PANT]) (Chen et al., 1997). Using the PANT approach, DNA damage has been described in a significant number of cells of the caudate putamen and in few cortical cells, starting at 4 hours after the onset of ischemia (Nagayama et al., 2000). Therefore, it seems that the comet assay has a higher sensitivity in detecting low levels of DNA damage because a significant increase of DNA damage was detected only 2 hours after the artery occlusion. In addition, when compared with histologic procedures, the comet assay offers the possibility of quantifying DNA damage for each analyzed cell, thus allowing evaluation of damage distribution within a given population of cells. Morphologic methods, however, allow further characterization of the DNA-damaged cells with immunohistochemistry, which obviously is not possible when using isolated nuclei. Single-strand breaks can also be detected by using DNA laddering under alkaline conditions (Specht et al., 2000). However, this procedure does not allow single cell resolution and requires substantial amounts of tissue.
Because the main focus of the present study was the early, free-radical-induced DNA damage, which consists mainly of SSBs, this investigation was conducted within 4 hours from ischemic insult. The neutral comet assay can be used to detect DSBs occurring during apoptotic cell death (Fairbairn et al., 1995), which, in the ischemic brain, takes place at later stages. For example, this approach has been used in human lymphocytes treated with bleomycin, and it has been demonstrated that the detection of DSBs with the neutral comet assay coincides with the presence of apoptosis evaluated with either morphologic or DNA-laddering approaches (Benitez-Bribiesca and Sanchez-Suarez, 1999). As already discussed herein for PANT, the comet assay can provide additional information for studying ischemia-induced apoptosis as compared with morphologic methods, such as terminal deoxynucleotidyl transferase-mediated dUTP labeling.
To make the assay suitable for CNS studies, we modified the originally described procedure (Singh et al., 1988) and performed the experiments in isolated nuclei. This strategy minimizes the manipulation of the tissue and reduces the time lag between the collection of the brain and the beginning of DNA break measurements. In fact, the procedures commonly used to acutely isolate brain cells require enzymatic treatments and hours to be completed (Stefani et al., 1996), whereas the isolation of nuclei consists of a simple homogenization in appropriate medium (Kaufmann and Shaper, 1984) and can be completed in minutes. Indeed, the number of DNA breaks we measured under basal conditions (i.e., in naive animals and in the contralateral side of animals that have undergone MCAO) was low, because most of the nuclei exhibited little or no electrophoretic migration. However, we also detected a small percentage of cells exhibiting higher levels of DNA damage under basal conditions. Similar results have been obtained in previous studies in human lymphocytes and in various mouse organs (Arlett et al., 1993; Collins et al., 1995; Sasaki et al., 1997), and it is possible that this background level of DNA damage is caused by the nuclei isolation procedure. Obviously, we cannot rule out the possibility that the SSBs we detected are present in vivo under control conditions.
It is worth noting that the amount of tissue required for each sample according to this procedure is approximately 2 mg (wet weight). This makes the method potentially useful for the study of DNA damage in much smaller brain areas than those chosen in the present study.
To further evaluate the use of the comet assay in the study of the early molecular events in ischemic brain, we used two different molecules, both able to experimentally reduce brain infarct after MCAO. The first was MK-801, an NMDA receptor antagonist, and the second was DPQ, a PARP inhibitor (Meldrum, 1985; Choi, 1988b). In our experiments MK-801 was able to reduce the early increase in DNA damage induced by MCAO, in agreement with the widely accepted notion that NMDA receptor stimulation concurs with generation and maintenance of a state of oxidative stress that in turn may induce damage to biologically relevant macromolecules, including DNA (Pellegrini-Giampietro et al., 1990; Huang et al., 1994; Love, 1999).
The PARP inhibitor DPQ has also been shown to exert neuroprotective actions during cerebral ischemia (Takahashi et al., 1999; Moroni et al., 2001). Because PARP activation is triggered by DNA damage, it is reasonable to assume that the mechanism of DPQ neuroprotective action involves a pathway located at a later stage in the cascade of events leading from the ischemic challenge to neuronal death (Zhang et al., 1994; Moroni et al., 2001). PARP inhibitors are supposed to achieve neuroprotection by preventing PARP overactivation, and reducing the cellular depletion of NAD and ATP (Lo et al., 1998; Takahashi and Greenberg, 1999). In agreement with this hypothesis, our results show that DPQ administration is effective in reducing the infarct volume, but not in preventing ischemia-induced DNA damage. Thus, these data suggest that protection from ischemic injury in the brain can be achieved with pharmacologic tools aimed at inhibiting stages of damage progression occurring downstream of excessive stimulation of glutamate receptors.
Finally, our observations show that the comet assay may be considered a sensitive method useful for studying DNA damage in brain pathology and neuroprotection.