
Abstract
Ischemic brain injury results in neuronal loss and associated neurologic deficits. Although there is some evidence that intercellular communication via gap junctions can spread oxidative cell injury, the possible role of gap-junctional communication in ischemia-induced cell death is the object of debate. Because gap junctions directly connect the cytoplasms of coupled cells, they offer a way to propagate stress signals from cell to cell. The authors investigated the contribution of gap-junctional communication to cell death using an in vitro ischemia model, which was reproduced by submersion of organotypic hippocampal slices into glucose-free deoxygenated medium. The gap-junctional blocker carbenoxolone significantly decreased the spread of cell death, as measured by propidium iodide staining, over a 48-hour period after the ischemic episode. Carbenoxolone ameliorated the hypoxia-induced impairment of the intrinsic neuronal electrophysiologic characteristics, as measured by whole-cell patch clamp recordings. To determine whether specific connexins were involved in the spread of postischemic cell death, the authors partially reduced the synthesis of specific connexins using antisense oligodeoxynucleotides. Simultaneous knockdown of two connexins localized mostly in neurons, connexins 32 and 26, resulted in significant neuroprotection 48 hours after the hypoxic– hypoglycemic episode. Similarly, partial reduction of the predominant glial connexin 43 significantly decreased cell death. These results indicate that gap-junctional communication contributes to the propagation of hypoxic injury and that specific gap junctions could be a novel target to reduce brain damage.
Interruption of cerebral blood supply results in severe reduction of cellular energy, followed by anoxic depolarization and impairment of ionic homeostasis (Siesjo, 1993; Dirnagl et al., 1999; Joshi and Andrew, 2001). The resulting accumulation of excitatory amino acids leads to a sustained N-methyl
Gap-junctional communication (GJC) is an important component in direct cell-cell communication that contributes to the maintenance of tissue homeostasis. The gap-junctional channels, formed by connexins, allow the passage of ions and small molecules (Spray and Dermietzel, 1996). Many connexins are expressed in the mammalian brain, and although their specific cellular localization remains controversial, it is generally accepted that connexins 26 (Cx26), 32 (Cx32), and 36 (Cx36) are synthesized in neurons (Dermietzel et al., 1989; Bruzzone et al., 1996; Nadarajah et al., 1997; Condorel li et al., 1998; Venance et al., 2000). Connexin 43 (Cx43) is mostly found in astrocytes, but is also present in pyramidal neurons (Nadarajah et al., 1996; Simbörger et al., 1997). When pathogenic factors accumulate in stressed cells, it is conceivable to propose that the spread of these factors from cell to cell via gap junctions may amplify the pathologic process, promoting the spread of damage. Observations indicating that GJC may play a role in spreading cell death include the ability of glial gap junctions to exacerbate cell death in dissociated cultures subjected to oxidative stress (Lin et al., 1998), and the reduction of injury in a rodent model of stroke by blocking GJC (Rawanduzy et al., 1997). Further evidence that GJC is critical for spreading cell death is the observation that the blockade of GJC with heptanol limited myocardial necrosis in cardiomyocytes (Garcia-Dorado et al., 1997), and that the passage of Na+ through gap junctions propagates cardiomyocyte hypercontracture after ischemic episodes, causing myocardial necrosis (Ruiz-Meana et al. 1999). Transmission of damage signals from unhealthy to normal cells via gap junctions has also been shown in a different pathology using α-particle irradiated cells (Azzam et al., 2001). In addition, other observations suggest that gap junctions are altered as a consequence of ischemic injuries. Ischemia-induced Cx43 dephosphorylation has been shown in heart (Huang et al., 1999) and brain tissue (Li et al., 1998; Beardslee et al., 2000). In cultures, Cx43 channels open during metabolic inhibition (John et al., 1999) and compromise the cell's ability to maintain ionic balance.
An important technical limitation in these studies is the lack of specific gap-junctional blockers. Most of the blockers used (octanol, halothane) interfere with excitatory synaptic transmission (Rorig et al., 1996; Puil et al., 1990; Puil and El-Beheiry, 1990; Pocock and Richards, 1993), an effect neuroprotective in itself. In this study, we sought to overcome these limitations and to explore whether specific gap junctions play a role in ischemic neurodegeneration. We used an in vitro model of ischemia (Frantseva et al., 1999) and present several lines of evidence that GJC promotes cell death in organotypic hippocampal slices over the 48-hour period after hypoxic–hypoglycemic insult.
MATERIALS AND METHODS
Preparation of organotypic slice cultures
Techniques for culturing brain slices were described previously (Perez Velazquez et al., 1997; Adamchik et al., 2000a). Briefly, the brains of 7-day-old male Wistar rats were aseptically removed and immersed in an ice-cold dissecting medium (pH 7.2) containing 50% minimal essential media without bicarbonate, 50% calcium- and magnesium-free balanced salt solution, 20-mmol/L HEPES, and 7.5-mmol/L
Hypoxic–hypoglycemic injury and reperfusion
The hypoxic submersion method was performed as described previously (Frantseva et al., 1999). Briefly, hippocampal organotypic slices were subjected (after 13–14 days in vitro) to the hypoxic-hypoglycemic (H–H) episode by submergence in 2.5 mL glucose-free deoxygenated (bubbled with 5% carbon dioxide and 95% nitrogen) culture medium for 1 hour, where glucose was substituted for by 10-mmol/L sucrose. The hypoxic and control slices were placed in the tissue culture incubator at 36°C to 37°C. After 1 hour, the slices were returned to conditions of unimpaired air-medium interface and the medium was replaced by the normal culture medium containing glucose. The oxygen partial pressure, pH (range, 7.2–7.4), and bicarbonate were measured in both oxygenated and ischemic medium (Stat Profile 10; Nova Biomedical, Waltham, MA, U.S.A.).
For electrophysiologic recordings, hypoxia–hypoglycemia was reproduced by superfusing slices (flow rate, 4–5 mL/min) with glucose-free deoxygenated artificial cerebrospinal fluid (ACSF) aerated with 95% nitrogen and 5% carbon dioxide; 10-mmol/L sucrose was added to the solution to maintain osmolarity. Glucose-free deoxygenated ACSF was applied for 8 minutes because it is not possible to maintain a stable whole-cell recording over a period longer than 1 hour. To monitor the time course of oxygen levels in the perfusion chamber, we used an oxygen probe (ISO2 Oxygen Meter; World Precision Instruments, Sarasota, FL, U.S.A.). As reported earlier (Perez-Velazquez et al., 1997), hypoxic conditions were achieved 1.5 to 2 minutes after the onset of perfusion with deoxygenated and glucose-free ACSF. It took a similar time to return to normoxic conditions while superfusing with normal ACSF.
Electrophysiologic recordings
For electrophysiologic recordings, slices were transferred to a superfusion chamber maintained at 36°C to 37°C (Model PDMI-2; Medical Systems, Saint Laurent, Quebec, Canada). The superfusion solution (ACSF) contained 125-mmol/L sodium chloride, 2.5-mmol/L potassium chloride, 1.25-mmol/L NaH2PO4, 2-mmol/L magnesium sulfate, 2-mmol/L calcium chloride, 25-mmol/L NaHCO3, and 10-mmol/L glucose (pH 7.4) when aerated with 95% oxygen and 5% carbon dioxide (305 ± 5 mOsm). Hypoxia–hypoglycemia was initiated by superfusing slices (flow rate, 4–5 mL/min) with glucose-free deoxygenated ACSF. For whole-cell electrical recordings, the internal solution in the patch recording electrode contained 150-mmol/L potassium gluconate, 10-mmol/L HEPES, 2-mmol/L magnesium-ATP, 5-mmol/L potassium chloride, and 0.1-mmol/L ethyleneglycoltetracetic acid (pH 7.2) adjusted with potassium hydroxide (275 ± 5 mOsm). Neuronal recordings were performed using the current clamp whole-cell configuration of the patch-clamp technique. Patch pipettes were pulled from borosilicate capillary tubing (World Precision Instruments). Electrodes had tip resistances ranging from 4 to 6 MΩ when filled with internal solution. The resistance to ground of the whole-cell seal was 2–8 Gil before breakthrough. Neuronal responses were recorded using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA, U.S.A.) with the low-pass filter setting at 1 kHz. PCLAMP software version 6.3 (Axon Instruments) was used for analysis of the membrane potential (Vm) and the input resistances (RN), which were measured from the linear part of the current-voltage plot. Data were stored using the PCLAMP software using a 12-bit D/A interface (Digidata 1200, Axon Instruments), or were filtered at 1 kHz, digitized at 88 kHz, and stored on video tape using a digital data recorder VR-10 (Instrutech, New York, NY, U.S.A.) for later playback and analysis. For extracellular field potential recordings, extracellular orthodromic electrical stimulation (100-μs pulse width) was delivered via a bipolar stimulating Teflon-insulated tungsten electrode, and an extracellular recording electrode was filled with ACSF and placed in the CA1 or CA3 cell body layers.
Antisense oligodeoxynucleotide treatment and Western blots
Antisense or missense oligodeoxynucleotides (ODNs) were obtained purified from Medicorp (Montreal, Canada). A total of eight antisense phosphorothioate oligonucleotides were tried for Cx26, Cx32, and Cx43, only three of which significantly reduced the level of the respective connexin protein, as judged by Western blot analysis (Fig. 4). The corresponding sequences are 5′–3′ antisense Cx32 GTATAGACCTGTCCAGTT, Cx43 ACTCCAGTCACCCAT, and Cx26 CTGTAGTGTGCCCCAATC; missense oligonucleotide, GTTTTAATTCCTAAG. To ensure the specificity of the sequences, a BLAST search (NIH Web site, www.ncbi.nlm.nih.gov/blast) was performed for each sequence. The ODNs were reconstituted in water to make a stock solution of 2 mmol/L, and then applied to cultures at 30 μmol/L using a 100-μL/mL liposomal preparation DOTAP (Boehringer, Mannheim, Germany) to improve delivery. Freshly diluted ODN was applied every 12, 24, or 48 hours. Optimal results were obtained if applied every 24 or 48 hours for Cx26 and Cx43, and every 48 hours for Cx32. For the experiments, antisense, missense, or vehicle was applied to the organotypic cultures every 48 hours, but did not result in any observable toxicity. We used phosphorothioate backbones in the ODNs because they confer more nuclease resistance. The liposomal preparation was used as it has been shown that using vehicles for delivery reduces the amount of oligomer needed, reduces nonsequence-specific interactions and protects against nuclease cleavage (Stein, 1998).
Western blotting was used to assess the efficacy of the antisense treatment at reducing the synthesis of specific connexin proteins. For Western blots, proteins were extracted with Triton X-100 from organotypic slices treated for 5 days with the oligomers as detailed previously. Protein concentration was estimated by the Lowry method (Sigma, St. Louis, MO, U.S.A.). Proteins were separated on a 12% polyacrylamide-sodium dodecyl sulfate gel and electrophoretically transferred to hydrophobic polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, U.S.A.). Blots were blocked for 60 minutes in 5% nonfat dry milk and 1% bovine serum albumin dissolved in TBST buffer (10-mmol/L Tris, 150-mmol/L sodium chloride, and 0.05% Tween 20) and incubated overnight in the presence of specific antibodies for Cx32, Cx26, or Cx43 (Calbiochem, San Diego, CA, U.S.A.). To ensure equality of loading between lanes (approximately 80 μg/lane), an anti–α-tubulin antibody (Calbiochem) was used at a concentration of 0.5 μg/mL to detect the tubulin protein band (Fig. 5). The blots were developed using the respective alkaline-phosphatase–labeled secondary antibodies (Promega, Madison, WI, U.S.A.). Densitometric analysis was performed using Quantity One software (Bio-Rad, Mississauga, ON, Canada). The outside diameters of the connexin bands were normalized to the α-tubulin band.
Determination of cell death
Organotypic slices were used after 13 or 14 days in vitro. Cell death was determined over a period of 48 hours after injury using the fluorescent viability indicator propidium iodide (PI), as described by Frantseva et al. (1999) and Perez Velazquez et al. (1999). The PI was applied at 10 μmol/L before the experiments in each dish. The PI fluorescence emission was measured immediately before and 24 and 48 hours after the ischemic insult using a laser scanning confocal microscope (BioRad MRC-600) and a 4x objective. A rhodamine filter (510–560/590 nm) was used to view PI fluorescence emission. Gains and black levels were standardized for each experiment. The fluorescence images were acquired and analyzed with the Comos and the Confocal Assistant software packages (BioRad). Pixel intensity measurements were averaged at the three main areas of the hippocampus, the cell body layers of CA1, CA3, or the dentate gyrus using a standard size box and the software features.
At the end of each experiment, slices were killed by submergence in ACSF for 48 hours at low temperature (4°C) in the presence of PI. The final PI fluorescence obtained after this treatment was considered to be the fluorescence that closely represented 100% cellular death. Cell death was then expressed as percentage of final fluorescence (Ffin) minus the background fluorescence (F0) taken before the injury, as shown by the equation % cell death = (Ft - F0)/(Ffin - F0) × 100, where Ft is the fluorescence of slices measured at several time points (normally 24 and 48 hours) after the onset of injury (Abdel-Hamid and Tymianski, 1997; Frantseva et al., 1999; Adamchik et al., 2000a). The statistical comparison between means was performed using analysis of variance and the unpaired Student's t-test, unless otherwise specified. Statistical significance was set at P < 0.05. Numerical values are expressed in the figures as mean ± SD.
Hippocampal slices exhibiting PI staining before ischemia or those revealing any incomplete or absent hippocampal layer were excluded from the experiments.
RESULTS
Effects of altering the strength of gap-junctional communication on the extent of cell loss after hypoxia–hypoglycemia
We use an in vitro ischemia-reperfusion model described previously (Frantseva et al., 1999). As shown in Fig. 1, our hypoxic insult results in selective neuronal loss, as judged by the preferential distribution of PI-positive nuclei in neuronal layers, where glial cells are sparse. Cell death was measured in three main neuronal hippocampal areas: CA1, CA2/3, and dentate gyrus (DG), by the established method that uses PI staining (Abdel-Hamid and Tymianski, 1997), and described in Materials and Methods. We also observed a reduction of evoked postsynaptic potentials during the reperfusion period (Frantseva et al., 1999). Hence, this in vitro model of ischemic injury is reliable and offers the opportunity to study long-term mechanisms of ischemic brain damage (Perez Velazquez et al., 1999). In preliminary studies, we observed dye coupling in organotypic hippocampal slices between neurons (one of four pyramidal cells filled with Lucifer yellow) and between glial cells (two of two; Frantseva et al., 2002; Perez Velazquez et al., 1996). Other studies have also shown dye coupling between neurons in organotypic slice preparations (Shinohara et al., 2000).
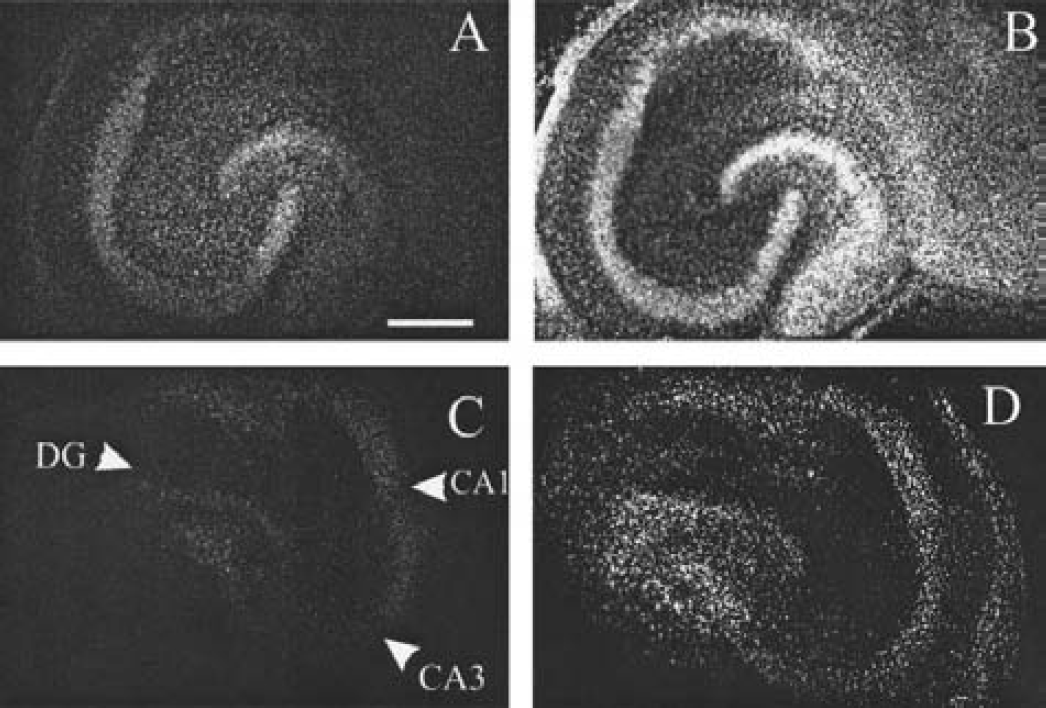
Cell death in organotypic hippocampal slice cultures after hypoxia–hypoglycemia is reduced by the presence of carbenoxolone. The slice is stained with PI to reveal cell death. (
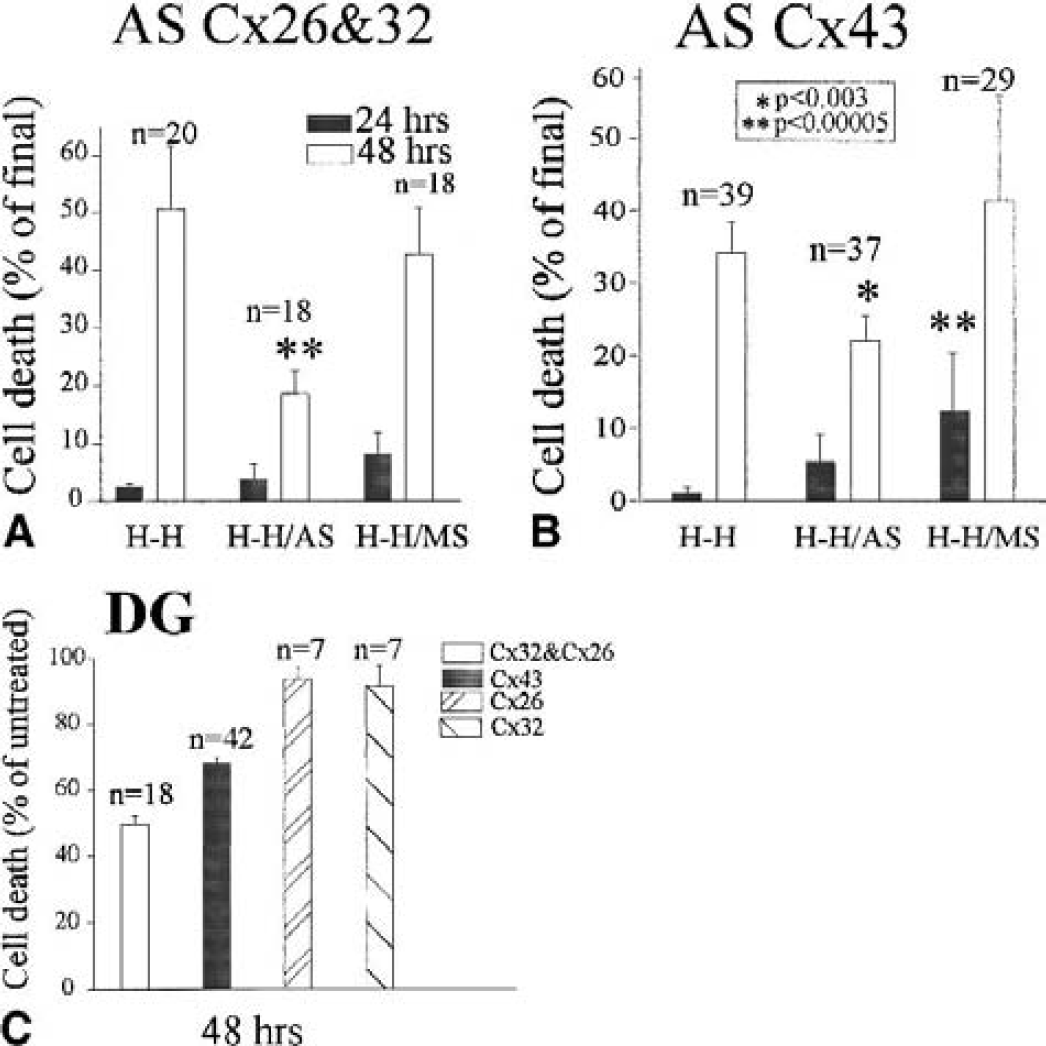
To determine whether GJC was partially responsible for the cell loss observed after our hypoxic–hypoglycemic episode, we used the gap-junctional blocker carbenoxolone, a derivative of glycyrrhetinic acid that has been very widely used to block gap junctions in several systems (Davidson and Baumgarten, 1988; Ishimatsu and Williams, 1996; Goldberg et al., 1996; Vaney et al., 1998; Bani-Yaghoub et al., 1999; Ross et al., 2000; Rozental et al., 2001; Traub et al., 2001; Schmitz et al., 2001). We showed in previous studies that carbenoxolone decreases dye coupling in organotypic slices (Frantseva et al., 2002). Incubating the organotypic cultures with carbenoxolone (120 to 150 μmol/L) resulted in significant neuroprotection (Figs. 1 and 2). The average cell loss in the pyramidal cell body layers (CA1, CA2/3) in the presence of carbenoxolone was 36 ± 9% of that found in untreated slices subjected to identical insults, and 48.2 ± 25% in the DG (these values represent averages of three experiments, n = 47 untreated and 35 treated slices). To obtain a time window for the neuroprotection observed, carbenoxolone was added at two time points after the injury, 2 and 24 hours after the hypoxic–hypoglycemic insult. Significant neuroprotection was observed if the blocker was added 2 hours after the ischemic episode, as shown in Fig. 2, though in this case cell death was higher than that found when the drug was present throughout the experiment, (average, 69.4 ± 5.8% of untreated injured slices measured 48 hours after injury in the pyramidal layers). In this case, no reduction in cell death was observed in the DG area (Fig. 2). However, addition of carbenoxolone 24 hours after injury did not result in appreciable neuroprotection in any hippocampal area. We should note that the concentrations of carbenoxolone used in these experiments (120 to 150 μmol/L) were nontoxic for our slices.
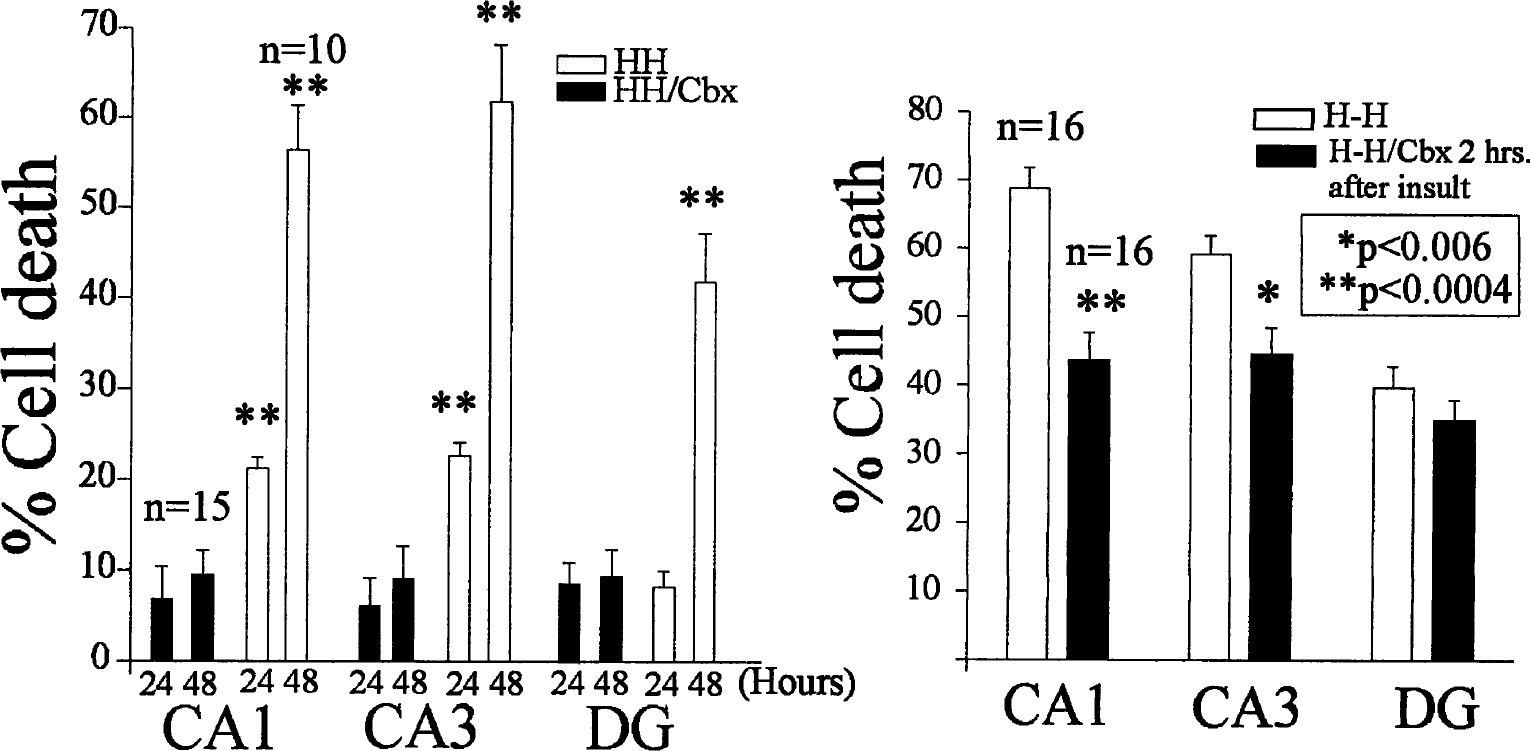
The gap-junctional blocker carbenoxolone (Cbx, 120 μmol/L) attenuates hypoxia-induced cell death in the three main hippocampal areas, CA1, CA3, and DG. The left graph represents the percentage of cell death (measured by PI fluorescence) at 24 and 48 hours after the hypoxic– hypoglycemic (HH) episode in nontreated (white bars) and treated slices (black bars). Cell death is still reduced if carbenoxolone is added 2 hours after the ischemic insult (black bars), at least in the pyramidal layers CA1 and CA3 (right graph). Cell death was determined at 48 hours. Asterisks denote statistical significance (unpaired Student's t-test) between injured nontreated and treated slices.
Effects of altering the strength of gap-junctional communication on the hypoxia-induced impairment of intrinsic neuronal characteristics
Long-term recordings revealed a progressive decrease in the extracellularly recorded evoked postsynaptic potentials over a 24 to 48 hour period (Frantseva et al., 1999). In parallel to this impairment of synaptic function, the neuronal intrinsic biophysical membrane properties were deteriorating during reperfusion, as reported previously. Briefly, whole-cell recordings from CA1 pyramidal neurons in the organotypic slices revealed that their resting membrane potentials (Vm) depolarized initially during the hypoxic–hypoglycemic insult, repolarized briefly at the onset of reperfusion, and continued depolarizing during reperfusion for the recording period (40–50 minutes after the ischemic episode) (Perez Velazquez et al., 1997, 2000). For these recording experiments, an 8-minute hypoxic–hypoglycemic insult was applied, because we were interested to observe the electrical characteristics in pyramidal cells using the patch-clamp method. Hence, the ischemic episode had to be of shorter duration (we cannot perform stable whole-cell recordings over 1.5 hours). The neuronal depolarization we observed during reperfusion could be the cellular correlate of the phenomenon termed spreading depression, a massive neuronal and glial depolarization known to follow ischemic (Leao, 1947; Nedergaard and Hansen, 1993; Obeidat and Andrew, 1998; Rawanduzy et al., 1997) and traumatic injuries (Katayama et al., 1990). The presence of carbenoxolone significantly reduced the irreversible neuronal depolarization during reperfusion (Fig. 3). Similarly, the hypoxia-induced decrease in the membrane input resistance (RN) was alleviated in slices incubated with the gap-junctional blocker (Fig. 3). A prolonged decrease in RN indicates a decline of the integrity of the plasma membrane. In fact, an average increase in RN values were observed during reperfusion in the presence of the drug.
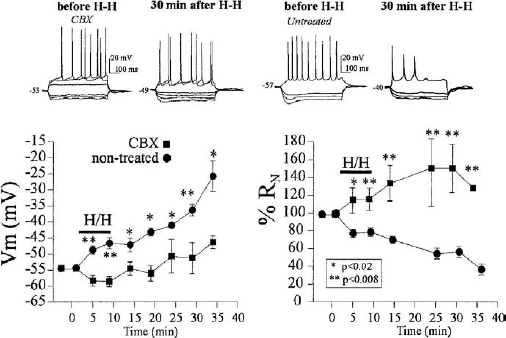
Carbenoxolone treatment alleviates the impairment of intrinsic electrophysiologic characteristics of CA1 neurons during ischemia-reperfusion. The CA1 neurons were whole-cell recorded and the average membrane potential (Vm, left graph) and input resistances (RN, right graph), measured during the hypoxic insult (H-H) and up to 30 minutes during reperfusion. Upper recordings depict voltage traces recorded from two pyramidal neurons in response to the same hyperpolarizing and depolarizing current pulses, before and 30 minutes after the ischemic insult. Their Vm values are shown in millivolts to the left of the traces. Notice that, without carbenoxolone (untreated, right traces), the cell depolarizes significantly and its RN decreases, quantified in the graphs below for several cells. The Vm in cells in untreated slices (n = 31) depolarize more than that of cells in slices pretreated with carbenoxolone (120 μmol/L, n = 11) for 2 to 3 hours before the insult (left graph). Relative changes in RN, taking the initial value as 100%, show that the RN values of cells pretreated with carbenoxolone do not decrease compared with nontreated cells. Indeed, an RN increase is seen during reperfusion (right graph). Asterisks denote statistical significance comparing values in treated and nontreated cells.
To assess possible electrophysiologic effects of carbenoxolone in the organotypic hippocampal slices, control noninjured slices were incubated with the drug and field potential and whole-cell recordings were obtained. No significant changes were observed. Specifically, field potential recordings in the CA1 cell body layer in slices that had been incubated with carbenoxolone (150 μmol/L) for 24 to 48 hours revealed no significant differences in the amplitude of the evoked population spikes or synaptic potentials (the extracellular stimulating electrode was positioned in the Schaffer collaterals). The average amplitudes were 105.2 ± 0.1% (postsynaptic potential) and 99.7 ± 0.1% (population spike, n = 7) of the amplitudes before carbenoxolone application. The lack of effects of carbenoxolone on the field postsynaptic potentials recorded in the pyramidal layers of the hippocampus was also found in other studies (Ross et al., 2000). Similarly, no significant alterations in Vm or RN were detected in whole-cell recordings from CA1 or CA3 pyramidal neurons, the average Vm was −54.8 ± 0.6 mV (n = 30) and mean RN was 102 ± 7 MΩ in control pyramidal neurons, and the respective values in slices incubated for 2 hours in the presence of carbenoxolone were −53.8 ± 0.9 mV (n = 10, P > 0.3 compared with the control group) and 100 ± 12.5 MΩ (P > 0.3). The lack of carbenoxolone effects on cell conductances, kinetics of stepped currents and intrinsic neuronal properties, has also been shown in other studies using brain slices and similar concentrations of the drug (Travagli et al., 1995; Osborne and Williams, 1996; Schmitz et al., 2001). Hence, the carbenoxolone effects reported in this study, maintaining normal intrinsic membrane characteristics during ischemia-reperfusion, cannot be attributed to nonspecific effects on the basic electrical neuronal properties or interference with synaptic function.
Hypoxia-induced cell loss is diminished in slices where connexin synthesis is reduced by specific antisense oligodeoxynucleotide treatment
An important limitation in the use of carbenoxolone (or any other gap-junctional blocker) is that it is not specific for a particular type of gap junction. Hence, we investigated whether specific connexins could be responsible for enhancing the cellular vulnerability to the damage induced by our hypoxic–hypoglycemic insult. It is widely accepted that hippocampal neurons synthesize Cx26, Cx32, Cx47, and most commonly Cx36 (Dermietzel et al., 1989; Bruzzone et al., 1996; Spray and Dermietzel, 1996; Nadarajah et al., 1996; Condorelli et al., 1998; Teubner et al., 2001), whereas Cx43 is mostly present in astrocytes. To study the possible differential contribution of different connexins to the cell damage observed after the insult, the synthesis of specific connexins was partially reduced using antisense ODNs. We focused in the two neuronal connexins that, at the developmental stage at which we use the organotypic slices, are probably most abundant, Cx32 and Cx26 (Dermietzel et al., 1989; Nadarajah et al., 1997), and the glial Cx43. The Western blots in Fig. 4 show that these three connexins are indeed present in our slices when the experiments are performed 12 to 16 days in vitro. Previous immunohistochemical studies revealed immunoreactivity associated with Cx32 and Cx26 in the pyramidal cell body layers of the organotypic slices, whereas Cx43 immunoreactivity was mostly localized to the nonneuronal areas (Adamchik et al., 2000b). The Western blots (Fig. 4) revealed that the antisense treatment reduced the particular connexin protein level to some extent (Table 1). We could not achieve a very complete reduction (> 90%), which would have been desirable. To assess whether the partial reduction in connexin expression and synthesis was functionally reflected, we determined in previous studies that dye coupling was partially reduced in the antisense-treated slices (Frantseva et al., 2002).
Reduction of connexin protein by antisense oligodeoxynucleotides

Values represent the percentage reduction compared with the control (untreated slices) connexin bands. The value for the Cx43 experiment using the missense ODN is negative because the analysis revealed more protein in that band compared with the control band. Densitometric analysis, averaged from 3 experiments (one is shown in Fig. 4), revealed that the O.D. of the connexin bands, normalized to the α-tubulin band and taking 100% the O.D. of the connexin band in the control lane (lane 1 in Fig. 4), was lower in antisense-treated slices than in the control or the missense-treated slices.
ODN, oligodeoxynucleotides.
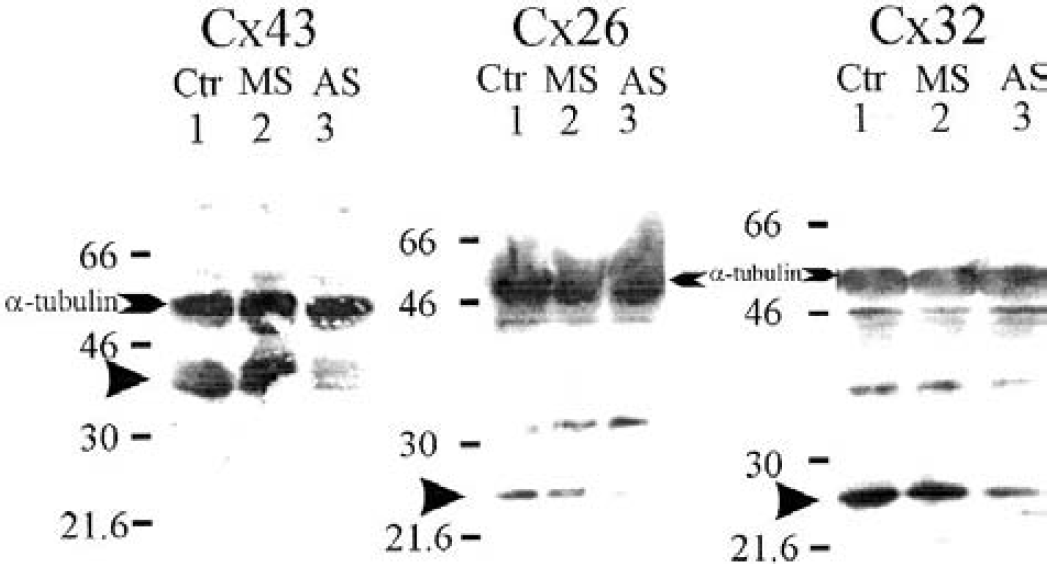
Western blots showing that treatment of organotypic hippocampal slices with antisense oligodeoxynucleotides (ODNs) reduce the amount of connexin protein. Slices were incubated with antisense (AS, lane 3), or missense (MS, lane 2) oligonucleotides (30 μmol/L) for Cx43, Cx26, and Cx32. Lane 1 (Ctr) was loaded with control samples from nontreated slices and incubated with the vehicle used to improve delivery of the ODNs. Numbers at left indicate the position of the molecular weight markers. α-Tubulin was used as the control protein to account for the amount of protein loaded in each gel lane (approximately 80 μg, Table 1). The Western blots were performed using antibodies against the specific connexin and another against tubulin. An arrow indicates the position of the band representing the specific connexin. Note that for Cx32, the band is at approximately 27 kd, a size observed by others and specified by the company.
The possible effects of the antisense-induced reduction in GJC on the extent of hypoxic cell death were then investigated. Organotypic hippocampal slices were subjected to the hypoxic–hypoglycemic episode and reperfused for 48 hours. Cell death was significantly less abundant in slices incubated in the presence of antisense ODNs for Cx26 and Cx32 simultaneously (Fig. 5), or Cx43 (Fig. 6). Quantification of cell death in these experiments is presented in Fig. 7. Treatment with antisense ODNs for Cx26 or Cx32 separately did not produce observable neuroprotection, cell death being defined as 105% of control untreated slices in the case of the treatment with ODNs for Cx26 (n = 19), and 94% of controls in the case of ODNs for Cx32 alone (n = 41). Cell death was also reduced in the DG area in slices treated with antisense for Cx43 and, more substantially, in coapplication of antisense ODNs for Cx32 and Cx26 (Fig. 7).
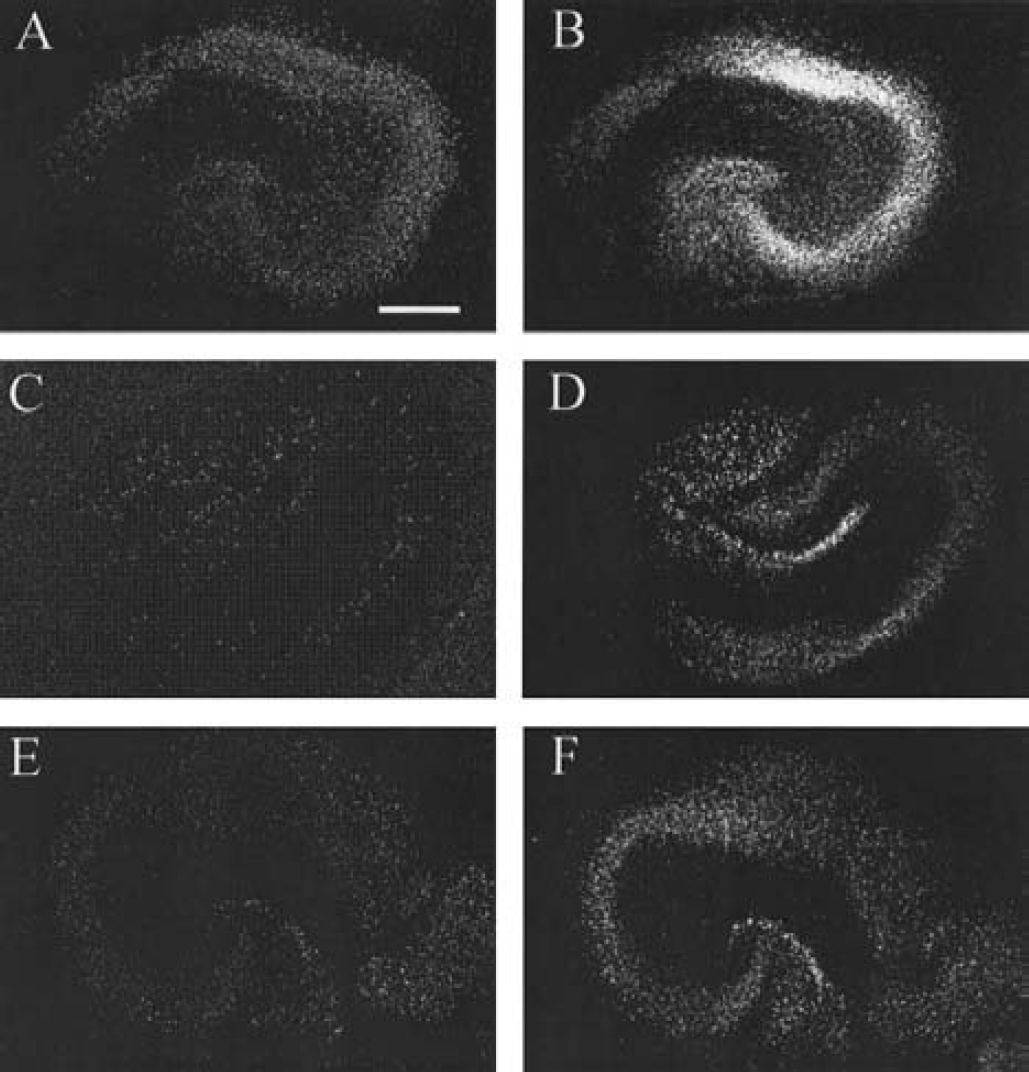
Reduction of GJC by antisense treatment results in decreased postischemic cell death. (
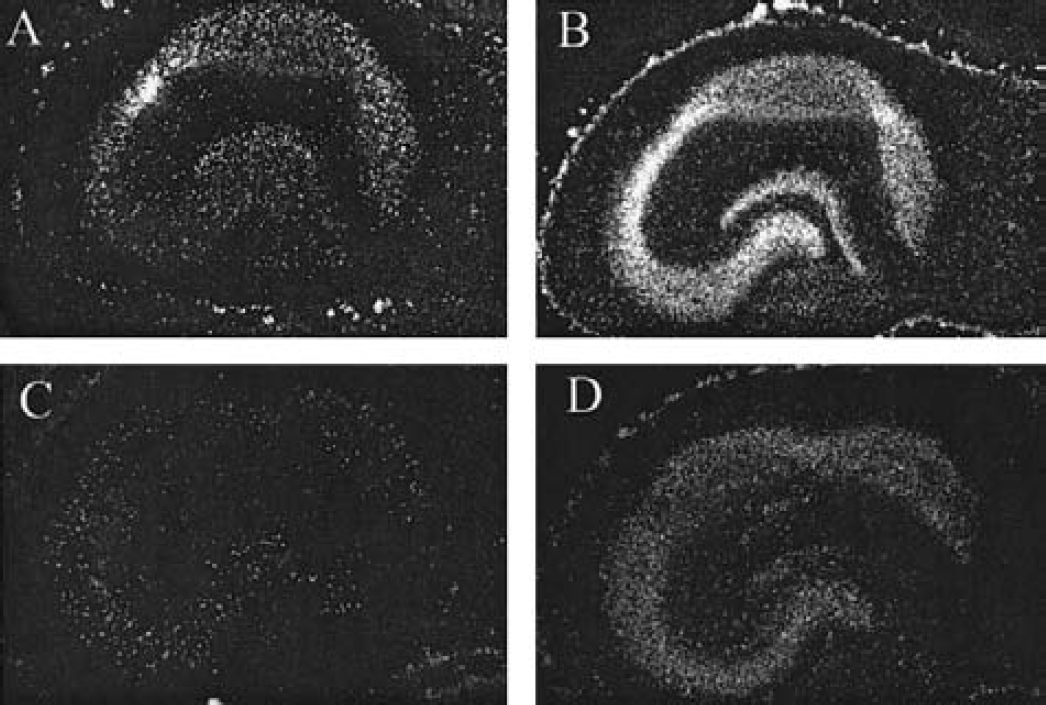
Treatment with antisense oligodeoxynucleotide (ODN) for Cx43 reduces the hypoxia-induced extent of cell death. (
Slices treated with antisense oligonucleotides for
These observations provide additional evidence for a role of specific gap junctions promoting hypoxia-induced cell death.
DISCUSSION
We have used an in vitro ischemia-reoxygenation model to study the contribution of GJC to the spread of hypoxic cell death. Our ischemia model results in selective neuronal death that can be measured by PI fluorescence emission at several time points in the same slice (Abdel-Hamid and Tymianski, 1997). Using a combination of pharmacologic manipulations to reduce GJC and molecular biological methods to reduce the synthesis of specific connexins, we present converging evidence for a role of GJC in enhancing the neuronal vulnerability to hypoxic injury.
The pathophysiology of ischemic injury consists of a phase of cell loss due to energy failure and loss of ionic homeostasis with overproduction of free radicals (Siesjo, 1993). Gap junctions could facilitate the spread of stress factors that accumulate during ischemia-reperfusion between coupled cells, as shown in the case of the transmission of damage signals in α-particle–irradiated coupled cells (Azzam et al., 2001) and in propagating apoptotic glial cell death after oxidative stress, calcium ionophores, or metabolic inhibition (Lin et al., 1998), insults closely related to ischemic conditions. The stress factors that may be spreading through the gap junctions may include sodium or calcium ions, apoptotic factors (Lin et al., 1998), lysophospholipids, or IP3, which is known to be involved in delayed cell death (Khan et al., 1996). Further recent evidence that GJC is critical for spreading cell death is the observation that by blocking gap junctions with octanol or halothane, the secondary expansion of infarction is reduced in rodent models of stroke (Rawanduzy et al., 1997; Saito et al., 1997). In addition, Garcia-Dorado et al. (1997) showed that the gap-junctional blocker heptanol limits myocardial necrosis in cardiomyocytes. Although these reports have provided interesting evidence for the involvement of GJC in spreading cell death, the blockers used (octanol and halothane) interfere importantly with synaptic transmission. Specifically, octanol reduces excitatory potentials in central neurons by at least 50% (Rorig et al., 1996), and halothane also attenuates excitatory glutamatergic and cholinergic inputs (Puil et al., 1990; Puil and El-Beheiry, 1990; Pocock and Richards, 1993), blocks spontaneous neuronal firing, and decreases spike amplitudes (Travagli et al., 1995). Because the reduction of excitatory input is known to be neuroprotective, it could be the case that the effects of these blockers are related to their inhibition of excitatory drive, and to less extent to the blockade of GJC. In this study, we used the gap-junctional blocker carbenoxolone (Davidson and Baumgarten, 1988; Goldberg et al., 1996; Bani-Yaghoub et al., 1999), which does not have significant interference with synaptic function in our system and in those of others (Ishimatsu and Williams, 1996; Ross et al., 2000), and does not significantly alter intrinsic neuronal characteristics (Travagli et al., 1995; Osborne and Williams, 1996; Schmitz et al., 2001).
The reduction of synaptic function during and after hypoxia–hypoglycemia in our system (Frantseva et al., 1999) and the neuronal depolarization that follows the hypoxic–hypoglycemic episode (Perez Velazquez et al., 1997, 2000) relate to the depressed neuronal activity found in other in vitro (Schiff and Somjen, 1987; Obeidat and Andrew, 1998; Joshi and Andrew, 2000; Kreisman et al., 2000) and in vivo (Leao, 1947; Nedergaard and Hansen, 1993; Rawanduzy et al., 1997) models. It is conceivable that the depolarization of neurons could represent the intracellular correlate of spreading depression (Leao, 1947; Martins-Ferreira et al., 2000), which involves a depolarization of large number of neurons and glial cells and has been proposed to determine the extent of postischemic cell death (Nedeergard, 1996; Obeidat and Andrew, 1998). Spreading depression is a common occurrence in other brain injuries, such as head trauma (Dixon et al., 1987; Bricolo and Turella, 1990; Katayama et al., 1990; Hovda et al., 1992). Considering that this phenomenon is associated with cell death if the tissue is metabolically compromised (Obeidat and Andrew, 1998) and that gap-junctional blockers arrest this depression and reduce infarct volume in an in vivo stroke model (Rawanduzy et al., 1997), it is possible that the progressive neuronal depolarization that we measured shortly after the onset of reperfusion could be involved in the delayed cell death. Along these lines of reasoning, carbenoxolone prevented the prolonged neuronal depolarization during reoxygenation in our whole-cell recordings, suggesting that this gap-junctional blocker might attenuate spreading depression, as found with other blockers (Rawanduzy et al., 1997). Indeed, carbenoxolone was not effective preventing cell death if administered 24 hours after the ischemic injury, a time when spreading depression may not be present because it normally occurs for a relatively short time.
It has been established that gap junctions remain open during brain ischemic conditions and uncoupling occurs because of the loss of membrane integrity (Cotrina et al., 1998). We could speculate that the transient alkalization reported to occur after ischemic episodes as a rebound from the acidification during the ischemia (Siesjo, 1993) enhances GJC, because gap junctions show a steep dependence on pH and the open states are favored by alkalization (Spray et al., 1981). Similarly, enhanced GJC after injuries has been shown in other systems after hyposmotic shocks in mouse astrocytes (Scemes and Spray, 1998) and after axotomy in motor neurons (Chang et al., 2000). Other studies showed that connexin channels open under metabolic restrictions similar to those that may occur during ischemia-reperfusion (John et al., 1999). Interestingly, an increase in glial GJC has been reported as a consequence of neuronal stimulation (Marrero and Orkand, 1996), an effect that may be occurring during hypoxia–hypoglycemia in our system, considering the neuronal hyperexcitability during the initial stages of the hypoxic episode (Perez Velazquez et al., 1997). Additional evidence for alterations of gap junctions during ischemia was revealed in studies using heart and brain tissue that have shown that Cx43 undergoes dephosphorylation after ischemia (Huang et al., 1999; Beardslee et al., 2000; Li et al., 1998). However, in dissociated cultures, astrocytic coupling is reduced 15 to 30 minutes after reoxygenation (Li and Nagy, 2000; Martinez and Saez, 2000). Also, Nishida et al. (2000) showed that reoxygenation causes a decrease in GJC between endothelial cells 2 hours after the onset of reperfusion. These discrepancies could be due to differences in model systems, cell types, or in the injuries administered.
One of the foremost problems in gap-junction research is that there are no connexin-specific blockers or drugs in general, and the manipulations to open or close gap junctions will affect all types (Rozental et al., 2001). In an attempt to determine whether specific connexins or gap-junctional pathways were involved in spreading ischemia-induced cell death, we reduced the synthesis of specific connexins using antisense ODNs. At the developmental stage at which we use our slices, both Cx26 and Cx32 are present in neurons (Dermietzel et al., 1989; Nadarajah et al., 1997; Adamchik et al., 2000b). Western blot analyses confirmed that these connexins were present in our slices. Therefore, it is not surprising that a simultaneous reduction in both connexins is needed to appreciate a neuroprotective effect because we did not find any effects when these connexins were partially knocked down separately. Also, diminishing glial connectivity (antisense for Cx43) was sufficient to reduce the hypoxia-induced cell loss, even though we should take into consideration that Cx43 has also been reported in pyramidal cells (Nadarajah et al., 1996; Simbörger et al., 1997). Because of the ubiquitous connexin expression, it is impossible to eliminate specifically interneuronal or glial coupling. These results indicate that a partial decrease in neuronal or glial GJC is enough to reduce the secondary damage.
The possible role of GJC in promoting or decreasing injury can find justification depending on the point of view. For example, GJC could be reasoned to be neuroprotective because glial cells will remove potassium or glutamate efficiently, and therefore neurons will not be subjected to large depolarizations with the consequent excitotoxicity (Blanc et al., 1998; Hansson et al., 2000). However, it can be reasoned that metabolic stress factors pass through gap junctions (Cotrina et al., 1998) and the wide spread of potassium and calcium waves through coupled astrocytes promotes release of glutamate from these cells, causing more excitotoxicity far from the focus. Which factor will predominate is hard to determine in a complex neuronal–glial network. It is entirely possible that, under some conditions, an enhanced GJC is neuroprotective, as evidenced in the case of ischemic insults to gastric mucosa (Iwata et al., 1998).
In summary, our data reveal a possible novel target to prevent the spread of hypoxia-induced injury. As shown in these studies, pharmacologic or molecular manipulations that reduce GJC significantly decrease the extent of posthypoxic cell death, even though none of these treatments causes a complete neuroprotection. Many other factors are obviously involved in spreading the injury (Choi, 1998; Dirnagl et al., 1999). We have now gathered evidence for the relative contribution of direct intercellular coupling. These observations may provide valuable information about possible therapeutic strategies to arrest the spread of the injury.