
Abstract
There is currently great interest in developing radiolabeled substrates for acetylcholinesterase and butyrylcholinesterase that would be useful in the in vivo imaging of patients with Alzheimer's disease. Using a simple in vitro spectrophotometric assay for determination of enzymatic cleavage rates, the structure-activity relationship for a short series of 1-methyl-4-piperidinyl esters was investigated. Relative enzymatic hydrolysis rates for the well-characterized 1-methyl-4-piperidinyl acetate, propionate, and i-butyrate esters were in agreement with literature values. The 4 and 5 carbon esters of 1-methyl-4-piperidinol were specific for butyrylcholinesterase and cleaved in the rank order n-valerate > n-butyrate >> 2-methylbutyrate, iso-valerate. These spectrophotometric results were also in agreement with in vitro hydrolysis rates in mouse blood and with in vivo regional retention of radioactivity in mouse brain of 11C-labeled analogs. Brain uptake and apparent enzymatic rate constants for 1-[11C]methyl-4-piperidinyl n-butyrate and n-valerate were calculated from in vivo measurements in M. nemistrina using positron emission tomography. Based on higher brain uptake of radioactivity and superior pharmacokinetics, 1-[11C]methyl-4-piperidinyl n-butyrate was identified as a new radiopharmaceutical for the in vivo measurement of butyrylcholinesterase activity.
Keywords
Acetylcholinesterase (AChE) [EC 3.1.1.7] is the hydrolytic enzyme responsible for metabolism of acetylcholine, leading to termination of cholinergic neurotransmission. Deficits in the AChE-mediated cleavage rate of [3H]acetylcholine (3H-ACh) have been observed in the histochemical analysis of postmortem brain samples from patients with Alzheimer's disease (AD) (Bierer et al., 1995). This observation, among others, has led to the hypothesis that the cognitive decline observed in AD is linked to progressive cholinergic degeneration.
In fact, the only treatment for AD currently available is the administration of AChE inhibitors, such as tacrine and donepezil (Taylor, 1998), which are thought to enhance cognition indirectly through elevation of acetylcholine concentrations. As a result of this correlation, there is currently a great interest in radiopharmaceuticals for the in vivo measurement of cholinergic neurochemistry. In fact, the AChE-mediated metabolism and trapping of radiopharmaceuticals for positron emission tomography (PET) based on AChE substrates is currently under clinical investigation as a direct in vivo index of AChE enzymatic activity in various brain regions (Koeppe et al., 1999; Kuhl et al., 1999, 2000; Namba et al., 1999; Shinotoh et al., 2000).
In addition to the decreases in AChE histochemical reactivity, changes in the related enzyme butyrylcholinesterase (BuChE) [EC 3.1.1.8] in postmortem analysis of AD brains have been reported (Perry et al., 1978; Huff et al., 1989; Mesulam and Geula, 1994). An approximate 10-fold increase in BuChE histochemical reactivity, as measured by cleavage of butyrylthiocholine, in regions of the temporal cortex was observed (Mesulam and Geula, 1994), with nearly all of this enzyme activity associated with amyloid plaques. In addition to these temporal increases in BuChE in AD, an increase in hippocampal BuChE associated with aging has been reported (Perry et al., 1978). In contrast, others have observed an approximate 4-fold increase in the ratio of 3H-ACh and [3H]butyrylthiocholine (3H-BCh) cleavage rates (3H-ACh/3H-BCh) in the caudate nucleus (Huff et al., 1989). It was not determined in this latter study whether the change in the enzyme ratio was because of an increase in AChE, a decrease in BuChE, or some combination of factors. In light of this controversy, it is of interest to develop a reliable in vivo method for measuring BuChE activity in various regions of the human brain.
The N-methylpiperidinyl esters have been extensively characterized as synthetic substrates for acetylcholinesterase (Irie et al., 1994, 1995, 1996; Namba et al., 1994; Kilbourn et al., 1996, 1998; Frey et al., 1997; Koeppe et al., 1999). Currently, 11C-labeled analogs of two of these esters, 1-[11C]methyl-4-piperidinyl acetate (AMP or MP4A) and 1-[11C]methyl-4-piperidinyl propionate (PMP or MP4P), are in routine clinical use as radiopharmaceuticals for studying Alzheimer's disease using PET (Iyo et al., 1997; Kuhl et al., 1999, 2000; Namba et al., 1999; Shinotoh et al., 2000). Both [11C]AMP and [11C]PMP are cleaved by AChE, and the labeled metabolite, 1-[11C]methyl-4-piperidinol, is trapped in the tissues. Kinetic analysis of this radioactivity trapping provides an estimate of the apparent rate constant for enzymatic hydrolysis that is proportional to the product of the actual rate constant for substrate cleavage and the enzyme concentration in the given region (Koeppe et al., 1999; Namba et al., 1999). Results from these studies have been very encouraging. Statistically significant localized decreases have been observed in the cortical hydrolysis rate of [11C]AMP or [11C]PMP in moderate AD versus age-matched controls (Kuhl et al., 1999; Shinotoh et al., 2000).
Also, [11C]PMP has been used to quantify AChE inhibition in vivo in rodents (Kilbourn et al., 1999), primates (Frey et al., 1997), and humans (Kuhl et al., 1999, 2000). These in vivo functional imaging methods will prove highly useful for investigating the changes in AChE associated with AD and will aid in evaluating the efficacy of new AChE inhibitors.
These early successes in the functional imaging of AChE and the possible clinical relevance of BuChE in AD have led the authors' laboratory to develop PET radiopharmaceuticals for measuring BuChE activity in vivo (Snyder et al., 2000). There is also interest in developing such AChE and BuChE substrates as radiopharmaceuticals for single photon emission computed tomography (SPECT), which will require significant structural modification of known enzyme substrates (Ueda et al., 1997, 1999, 2000; Yomoda et al., 1997, 1999).
To expedite initial screening of new radiotracer candidates, the authors have developed a simple in vitro spectrophotometric method for determining enzyme-mediated hydrolysis rate, relative to known cholinesterase substrates. This in vitro method will minimize the radiochemistry and in vivo evaluations necessary for identification of promising new radiotracers. Using this method, the authors performed a structure-activity relationship study of several N-methyl-4-piperidinyl esters to determine the effect of ester length and branching on the relative rates of enzyme-mediated hydrolysis by AChE and BuChE. Spectrophotometric data were compared with radiotracer metabolism in whole blood measured in vitro. These in vitro results then were used to direct in vivo biodistribution studies in mice and PET imaging studies in primates.
MATERIALS AND METHODS
Chemistry
The preparations of 1-methyl-4-piperidinyl acetate, propionate, and iso-butyrate, as well as 4-piperidinyl acetate and propionate, have been reported previously (Irie et al., 1994; Bormans et al., 1996; Snyder et al., 1998). All new compounds were characterized by melting point, 1H-NMR, and high-resolution mass spectrometry. A summary of these results is shown in Table 1.
Chemical structures, acronyms, and analytical data for the 4-piperidinyl esters
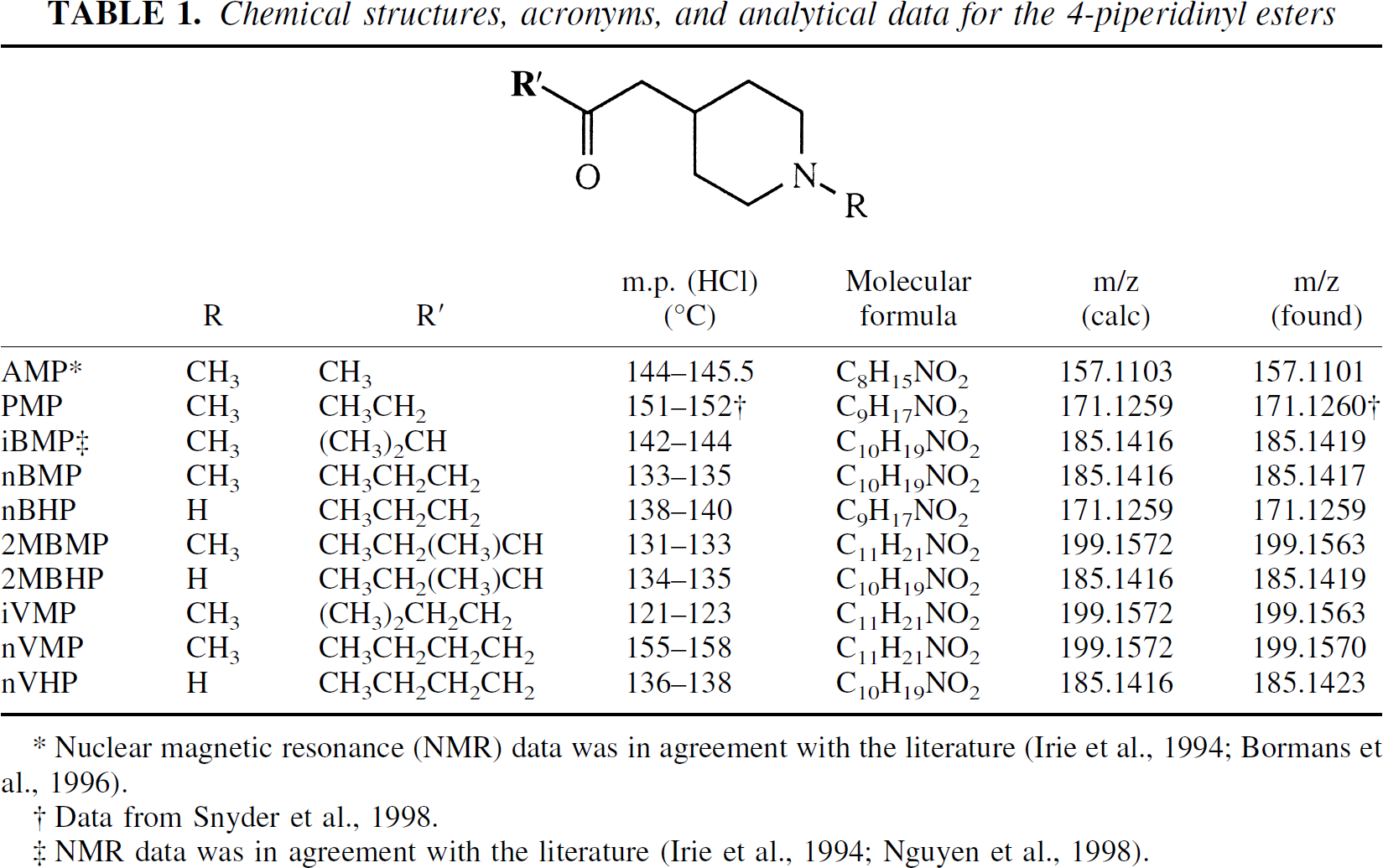
Nuclear magnetic resonance (NMR) data was in agreement with the literature (Irie et al., 1994; Bormans et al., 1996).
Data from Snyder et al., 1998.
NMR data was in agreement with the literature (Irie et al., 1994; Nguyen et al., 1998).
Electron ionization mass spectra were obtained on a V.G. Analytical 70-250S spectrometer (Micromass, Manchester, U.K.) and high-resolution spectra were within 0.001 m/z (10 ppm) unless otherwise indicated. Melting point determinations were performed on a Mel-Temp apparatus (Laboratory Devices, Cambridge, MA, U.S.A.) and are uncorrected. All starting materials for chemical syntheses were purchased from Aldrich Chemical (Milwaukee, WI, U.S.A.). Chromatography columns for high performance liquid chromatography (HPLC) analyses and purification were purchased from Phenomenex (Torrance, CA, U.S.A.). Supporting data for all syntheses are available upon request.
4-Piperidinyl esters
All piperidinyl esters used in radiolabeling reactions, except the 2-methylbutyrate ester, were prepared using the method reported previously for 4-piperidinyl propionate (Snyder et al., 1998), condensing 1-benzylcarbamoyl-4-hydroxypiperidine with the appropriate acyl chloride. All acyl chlorides used in these preparations are commercially available. The 2-methyl butyrate ester was prepared by coupling of 1-benzylcarbamoyl-4-hydroxypiperidine with 2-methylbutyric acid in the presence of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (Dhaon et al., 1982). In all cases, removal of the benzylcarbamate protecting group was accomplished by hydrogenolysis at atmospheric pressure over 10% Palladium on activated carbon (Snyder et al., 1998). The reaction mixture was filtered through Celite, acidified with 1.0 mol/L anhydrous HCl in diethyl ether and evaporated to dryness. Recrystallization from acetonitrile/ethyl acetate provided the 4-piperidinyl ester hydrochlorides.
1-Methyl-4-piperidinyl esters
The N-methylpiperidinyl esters were prepared directly by acylation of 1-methyl-4-piperidinol in dichloromethane by the published method (Snyder et al., 1998). All acyl chlorides used in these preparations are commercially available except the 2-methylbutyryl chloride that was prepared from 2-methylbutyric acid (Desai et al., 1970). The solvent and excess acyl chloride then were removed through rotary evaporation and the residue was recrystallized from acetonitrile and ethyl acetate to provide the desired product as the hydrochloride salt.
1-[11C]Methyl-4-piperidinyl esters
Using the published procedure for preparation of [11C]PMP (Snyder et al., 1998), the 4-piperidinyl ester (0.8 to 1.0 mg), dissolved as the free amine in 100 μL of dry N,N-dimethylformamide (DMF), was methylated using [11C]methyl trifluoromethanesulfonate (Jewett, 1992). The reaction mixture then was diluted with 400 μL HPLC eluent (0.1 mol/L ammonium acetate in 15:85 ethanol/water, pH = 5.5) and injected onto a C8 reversed phase HPLC column for purification (Ultremex; 4.6 × 250 mm, 1.5 mL/min, t
In vitro hydrolysis rate determination
Purified enzymes.
Purified AChE [EC 3.1.1.7] (electric eel) and BuChE [EC 3.1.1.8] (horse serum) enzyme preparations were purchased from Sigma (St. Louis, MO, U.S.A.). Stock solutions of AChE or BuChE in sterile isotonic saline were prepared at a concentration of approximately 100 enzyme units (U) per milliliter and stored at 4°C. These stock solutions were tested daily for stability and found to maintain a constant enzyme activity for 4 to 5 days. Fresh serial dilutions of stocks to lower enzyme concentrations (1, 2, 10, and 20 U/mL) were prepared for each substrate assayed and were stored on ice during assays. Substrate solutions were prepared using 50 mg/mL ester hydrochloride in sterile isotonic saline. Rates of ester hydrolysis by AChE and BuChE were determined with a commercially available cholinesterase assay kit (Sigma) using the procedure of Rappaport et al. (1959), modified as follows.
Spectrophotometric assays.
Into each of 2 glass vials (8 mL) was placed 100 μL of active enzyme solution and 100 μL of 0.15 mol/L saline. The enzyme in one of these vials was inactivated by heating the sealed vial in a 65 °C water bath for 15 minutes. This inactivated enzyme then was used as the blank for spectrophotometric assays. To both the active (sample) and inactive (blank) enzyme solutions was added 1.5 mL distilled water, 1.0 mL m-nitrophenol (MNP, 0.75 g/L in phosphate buffer, pH 7.8; Sigma), and 100 μL substrate solution. The resulting assay mixtures were quickly transferred to 1-cm quartz cuvettes and placed in a Perkin-Elmer Lambda 20 spectrophotometer, equipped with a water jacketed temperature controller set at 25°C ± 0.1 °C. The lower recommended limit for reliable measurement of absorbance changes (ΔA) in this assay is reported as 0.05 absorbance units (Sigma product insert) and the sensitivity limit of the instrument is ΔA = 0.03. Therefore, after allowing 10 minutes for temperature equilibration, the absorbance measurement at 420 nm was recorded at 6-second intervals for 15 minutes or until ΔA > 0.05 (Fig. 1). Total assay duration, including equilibration time, ranged from 25 to 120 minutes. Data were analyzed using the Kinlab software package (Perkin-Elmer) to calculate the slope of the absorbance change relative to enzyme concentration (ΔA/min/U/L). Linearity of the absorbance readings over the assay period was verified using linear least-squares regression analysis (r2 > 0.98).
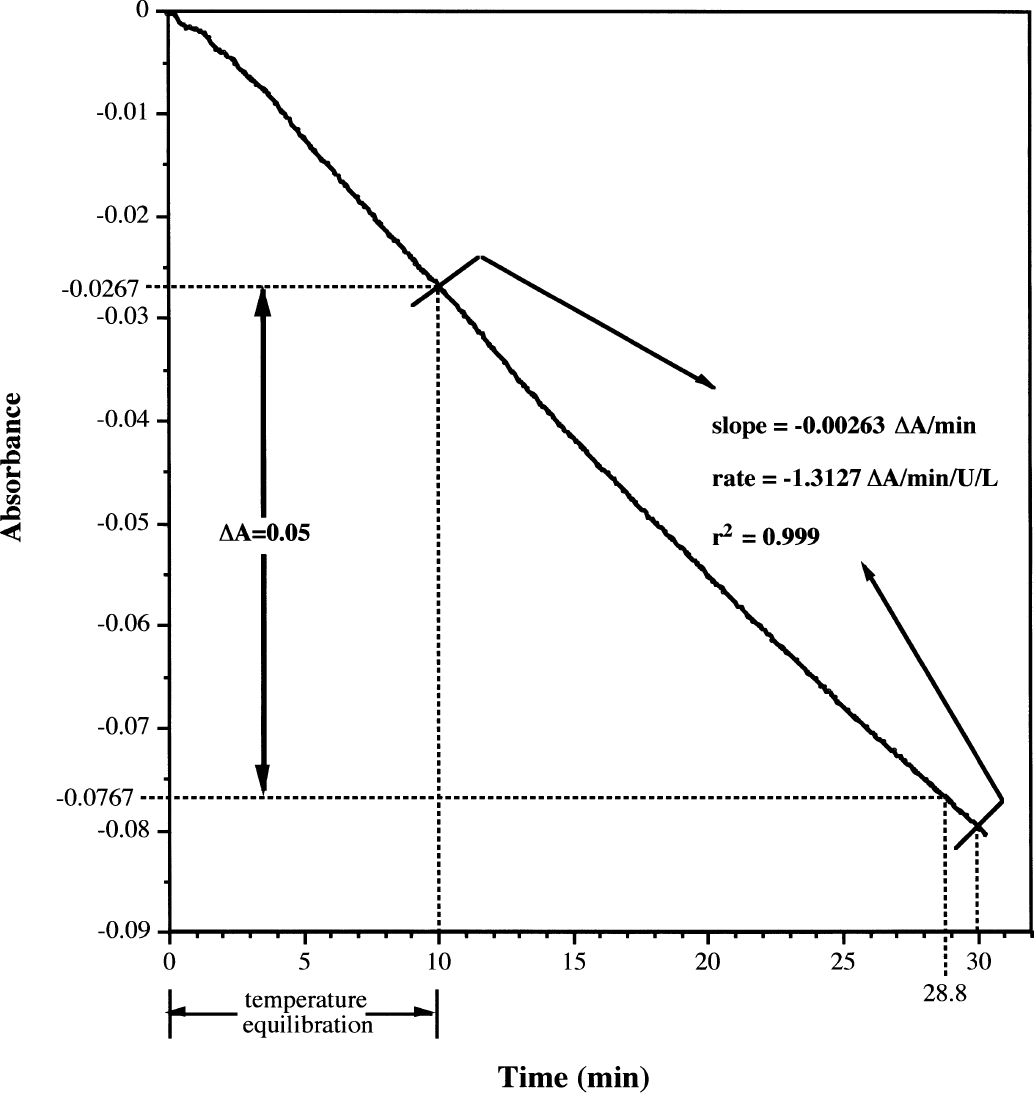
Typical spectrophotometric assay results exemplified by acetylcholine (ACh). Graph represents the absorbance at 420 nm over time for the cleavage of ACh (50 mg/mL) by acetylcholinesterase (AChE; 2 U/mL) in the presence of m-nitrophenol at 25 ± 0.1°C. Data are collected at 6-second intervals and 10 minutes is allowed for temperature equilibration before the start of slope calculation. Slope of the absorbance change is calculated from 10 minutes to time X, where the absorbance change (ΔA = A10 – Ax) is >0.05 absorbance units. The linearity of ΔA between 10 and × minutes was determined by linear least-squares regression (r2 > 0.98). The specific cleavage rate is calculated as the following: slope (ΔA/min) divided by the enzyme concentration ([AchE] in U/mL) then multiplied by 1000 mL/L. For this example, ΔA = 0.05 is achieved at 28.8 minutes so slope and linearity are calculated over the range of 10 to 30 minutes. Rate = −0.00263/2 (U/mL) × 1000 (mL/L) = −1.31 ΔA/min/U/L.
Mouse blood samples.
Samples of blood were harvested from 3 female CD-1 mice by cardiac puncture within 10 to 15 minutes before the start of metabolic rate determinations and were stored in a sealed heparinized sample tube (total sample volume typically ∼3 mL). Rate determinations were performed by placing approximately 1.2 mL of the whole blood in a 1.5 mL microcentrifuge tube, adding 200 μL (typically 2 to 4 mCi) 11C-labeled substrate in isotonic saline, and shaking the capped tube by hand to ensure complete mixing. The mixture then was allowed to stand at room temperature and 100 μL aliquots were removed at various time points between 1 and 60 minutes. These aliquots were quickly added to another microcentrifuge tube containing 500 μL absolute ethanol and mixed well to stop hydrolysis. The ethanol mixture then was centrifuged (1200 g for 1.5 minutes) and the supernatant was analyzed by thin-layer chromatography (silica, dichloromethane/methanol 95:5, NH3), with unlabeled ester and 1-methyl-4-piperidinol co-spots included for identification. Radioactivity on the thin-layer chromatography plates was visualized using a Fuji BAS-1800 phos-phorimaging system (Fuji Medical Systems, Stamford, CT, U.S.A.) and unlabeled spots were visualized with I2.
Regions of interest corresponding to the authentic radiotracer, 1-[11C]methyl-4-piperidinol, and chromatographic origin were drawn by hand on these images. An additional region of interest was drawn in a blank area of the plate for use in background correction, and the optical density for each region of interest was quantified. No other radioactive spots were observed. The fraction of authentic radiotracer at each time point was calculated as the optical density of the authentic tracer region of interest divided by the total signal in all regions and was converted to a percentage. A control sample for each experiment was prepared by adding 100 μL whole blood to 500 μL ethanol to destroy enzymatic activity, then adding 10 to 20 μL 11C-labeled substrate and analyzing the sample as described. Typically, control samples showed >98% authentic radiotracer with the remainder of the radioactivity trapped at the chromatographic origin. No metabolite was visible in any of these control samples.
In vivo regional distribution in mice
Female CD-1 mice (20 to 25 g, Charles River Laboratories, Wilmington, MA, U.S.A.) were anesthetized with diethyl ether and 150 to 250 μCi of the radiotracer, formulated in sterile isotonic saline, was injected through the tail vein. At 1, 5, 10, 20, 30, 45, and 60 minutes postinjection of radiotracer, groups of animals (n = 4) were killed by decapitation and the brain was rapidly removed and dissected into samples of striatum, cortex, hippocampus, thalamus, hypothalamus, cerebellum, pons-medulla, and remainder of brain. Tissue samples were weighed and the radioactivity counted in a Packard Autogamma 3780 automatic γ-counter (Packard Instrument, Meriden, CT, U.S.A.). The amount of radioactivity in each brain region was calculated as percentage of injected dose/g wet weight of tissue. Radioactivity retention fractions were calculated from the 1 and 30 minute data for selected brain regions (striatum, cortex, hippocampus, thalamus, and cerebellum), according to the published procedure (Kilbourn et al., 1999). Differences between regions in the amount of radioactivity retained were compared using paired Student's t-tests (P < 0.05 was considered significant). Differences in regional retention fractions between radiotracers were compared using unpaired Student's t-tests (P < 0.05 was considered significant).
Positron emission tomography imaging of primates
A typical primate imaging protocol is described in detail elsewhere (Kilbourn et al., 1996). Briefly, a female pigtail monkey (M. nemistrina, 4.6 kg) was anesthetized (ketamine and xylazine) and secured on the gantry of a TCC PC4600a tomograph (three-ring, five-slice tomograph); this PET scanner had been modified by addition of custom-fabricated lead collimators to enhance spatial resolution (inplane, 7.5 mm full width at half maximum). The animal then was injected with 20 to 30 mCi of radiotracer and imaged for 60 minutes. Analysis of these image data by methods described by Frey (Frey et al., 1997) and Koeppe (Koeppe et al., 1999) provided an estimate of the apparent rate constant for enzymatic hydrolysis (k3), which is the product of the actual rate constant for the reaction (khyd) and enzyme concentration ([ChE]) in the given region. The magnitude of k3 in the various brain regions is an index of local ChE activity (Koeppe et al., 1999).
RESULTS
In vitro ester cleavage rate: purified enzymes
The in vitro enzyme-mediated cleavage rate for each ester, using purified enzymes, was measured by incubating an excess of the substrate with a known concentration of either AChE (electric eel) or BuChE (horse serum), at 25°C, in the presence of the pH indicator m-nitrophenol (MNP) (Rappaport et al., 1959). Typical assay results are shown in Fig. 1. Ester hydrolysis releases a carboxylic acid into the reaction medium, resulting in a decrease in pH and hence an absorbance change in the indicator. In the presence of excess substrate, the slope of this MNP absorbance change is proportional to the maximum specific rate of ester hydrolysis for a given enzyme concentration (ΔA/min/U). The substrate concentration for all assays was fixed at 50 mg/mL.
The AChE cleavage rates for the five well-characterized ChE substrates—acetylcholine (ACh), butyrylcholine (BCh), 1-methyl-4-piperidinyl acetate (AMP), propionate (PMP), and iso-butyrate (iBMP), as well as 1-methyl-4-piperidinyl n-butyrate (nBMP)—are shown in Table 2. The cleavage rates by BuChE for all substrates examined—including 1-methyl-4-piperidinyl 2-methylbutyrate (2MBMP), iso-valerate (iVMP), and n-valerate (nVMP)—are shown in Table 3.
AChE-mediated cleavage rates for ChE substrates
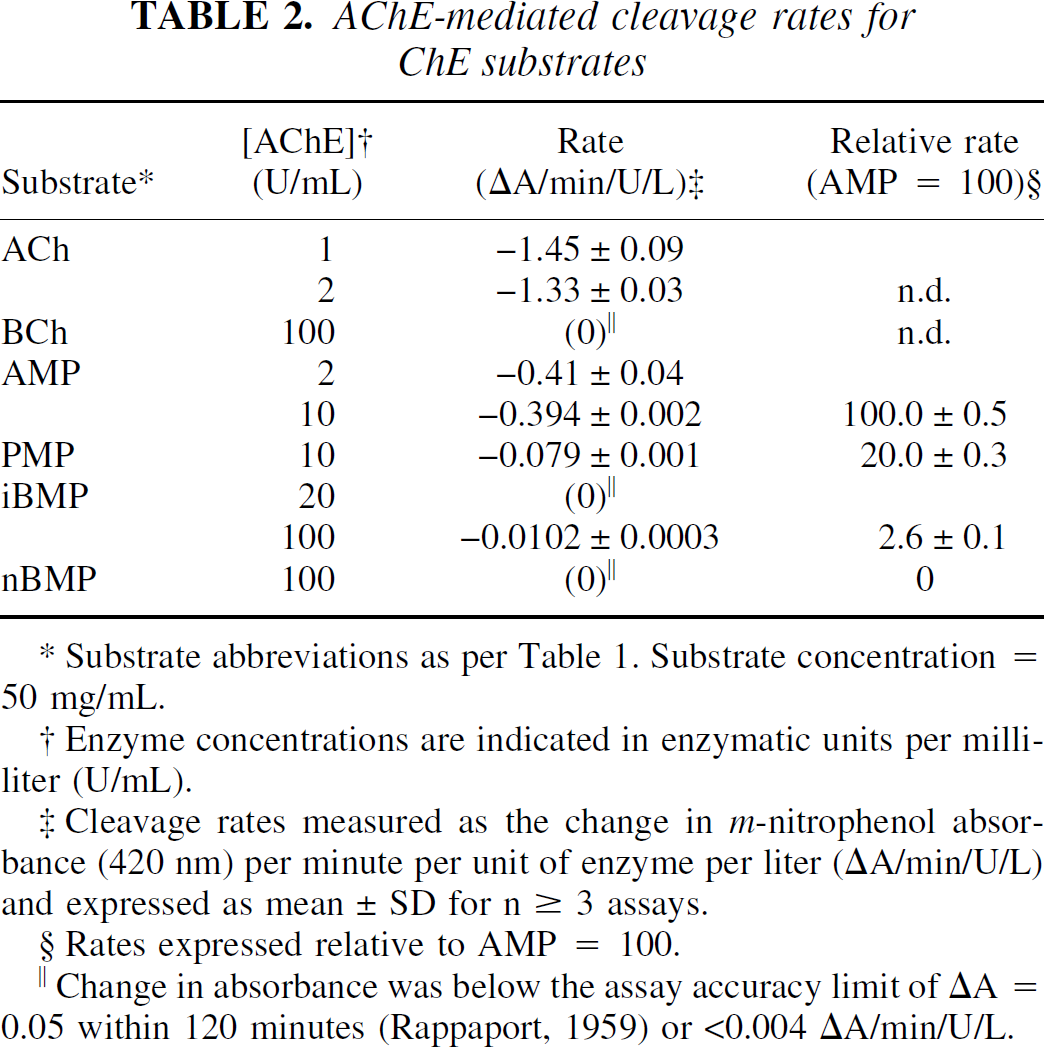
Substrate abbreviations as per Table 1. Substrate concentration = 50 mg/mL.
Enzyme concentrations are indicated in enzymatic units per milliliter (U/mL).
Cleavage rates measured as the change in m-nitrophenol absorbance (420 nm) per minute per unit of enzyme per liter (ΔA/min/U/L) and expressed as mean ± SD for n ≥ 3 assays.
Rates expressed relative to AMP = 100.
Change in absorbance was below the assay accuracy limit of ΔA = 0.05 within 120 minutes (Rappaport, 1959) or <0.004 ΔA/min/U/L.
BuChE-mediated cleavage rates for all ChE substrates
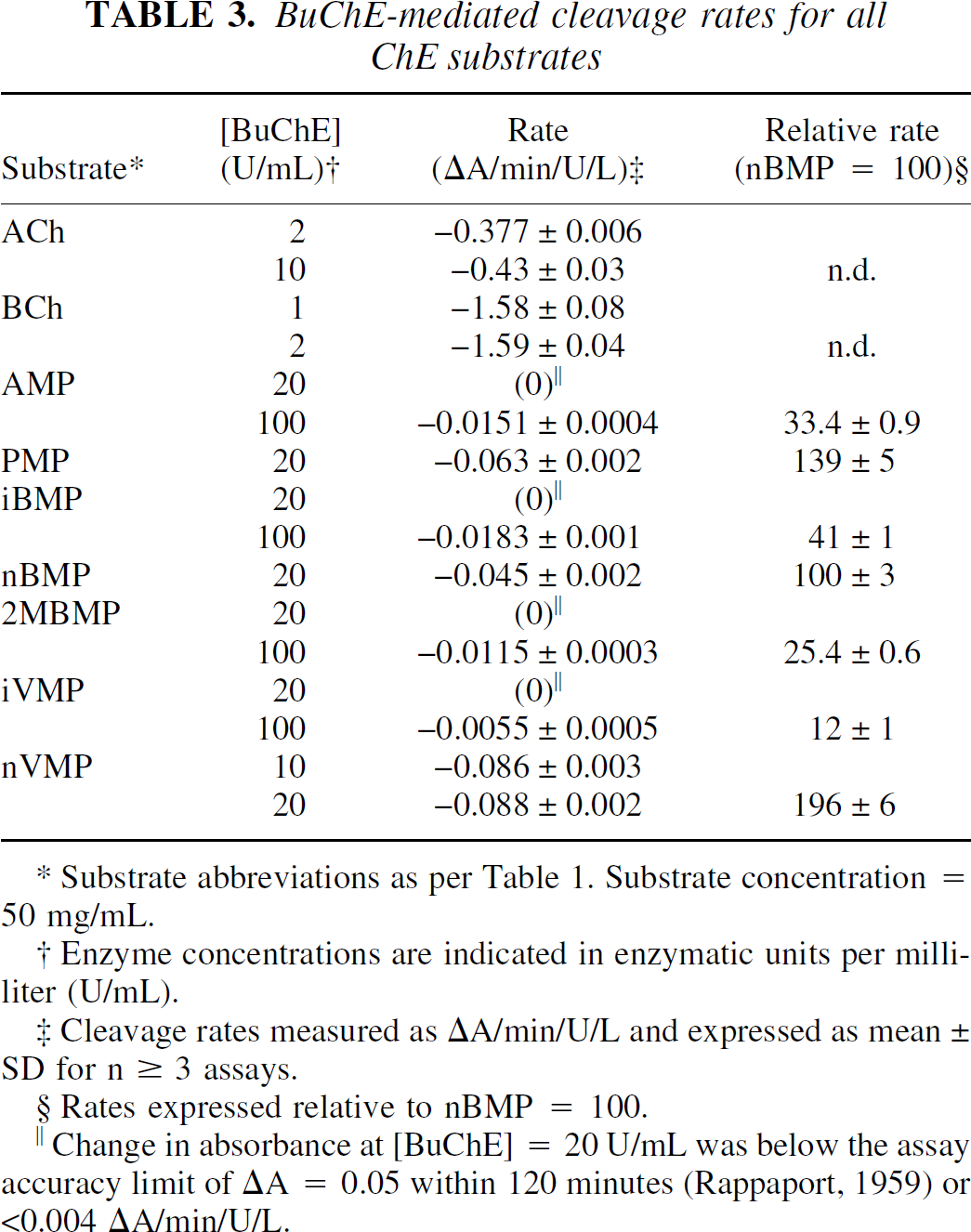
Substrate abbreviations as per Table 1. Substrate concentration = 50 mg/mL.
Enzyme concentrations are indicated in enzymatic units per milliliter (U/mL).
Cleavage rates measured as ΔA/min/U/L and expressed as mean ± SD for n ≥ 3 assays.
Rates expressed relative to nBMP = 100.
Change in absorbance at [BuChE] = 20 U/mL was below the assay accuracy limit of ΔA = 0.05 within 120 minutes (Rappaport, 1959) or <0.004 ΔA/min/U/L.
These data encompass a 100-fold range of cleavage rates from the rapidly hydrolyzed native substrates, ACh and BCh, to the most slowly reacting piperidinyl esters, such as iBMP or iVMP. It proved impossible to run all substrates at a common enzyme concentration, because at high enzyme concentrations the more rapidly hydrolyzed substrates (for example, ACh, BCh, and AMP) gave nonlinear absorbance changes resulting in unreliable rate values. Conversely, assay of a slowly hydrolyzed substrate, such as iBMP, at the lower enzyme concentrations used for the native substrates would be prohibitively lengthy (10 hours per assay) and would risk decomposition of the enzyme during the assay. Also, all of the cholinesterase substrates are somewhat labile over long periods in aqueous solution. For these reasons, the specific rate of cleavage per unit of enzyme (ΔA/min/U/L) for several substrates was measured at two different enzyme concentrations (Tables 2 and 3) and the values were compared using an unpaired Student's t-test. Under these conditions, none of the substrates exhibited specific cleavage rates with statistically significant (P < 0.05) enzyme concentration dependence and all ΔA rate measurements were linear during the course of the assay (r2 > 0.98).
These results confirm the validity of rate comparisons between substrates assayed at different enzyme concentrations (for example, AMP in 2 U/mL AChE versus iBMP in 100 U/mL AChE). However, it is possible that the rates determined for ACh and BCh are underestimated. Under the conditions used in the authors' assay, the substrate concentrations for the fastest reactions observed (ACh in 2 U/mL AChE and BCh in 2 U/mL BuChE) change approximately 15% during the measurement period. The absorbance measurements for these assays were still linear (r2 > 0.98), however, it is interesting to note that the largest rate discrepancies between measurements at different enzyme concentrations were seen for the cleavage of ACh (AChE: 1 U/mL vs. 2 U/mL, P = 0.069; BuChE: 2 U/mL vs. 10 U/mL, P = 0.052). Therefore, the AChE cleavage rates for the piperidine esters in Table 2 have also been expressed relative to AMP, rather than normalizing to the native substrate ACh as was done in previous studies (Snyder et al., 2000). Similarly, the BuChE cleavage rates for the piperidine esters in Table 3 have been expressed relative to nBMP.
The choice of AMP and nBMP for normalization was based on these compounds containing the analogous acetate and n-butyrate esters, respectively, found in ACh and BCh. Also, the cleavage rates for AMP and nBMP were much closer to those of the other piperidinyl esters examined. Normalization to these compounds avoided dependence on the possibly less accurate ACh and BCh data and provided larger relative rate differences between the compounds assayed.
In analyzing these spectrophotometric data it is very important to note that 1 U of AChE is defined as the amount of enzyme necessary to hydrolyze 1.0 μmol ACh/min at 25°C and pH 7.4. One unit of BuChE is similarly defined, but is based on the hydrolysis of BCh. Also, the enzymes used here are from different species: AChE is from electric eel and BuChE is from horse serum. Because of these differences in unit definition and source species, no direct comparison of AChE and BuChE ester cleavage rates is possible for a single substrate.
As expected from previous experiments (Vellom et al., 1993; Irie et al., 1994,1996; Snyder et al., 2000), none of the esters examined was specific for AChE. Both AMP and PMP were cleaved rapidly by AChE (Table 2) with an acetate:propionate ratio of approximately 5:1. This is consistent with the relative cleavage rates in mouse brain homogenates (acetate:propionate = 4.5:1) reported for the 14C-labeled esters (Irie et al., 1994). BCh and all of the longer chain piperidinyl esters (nBMP, 2MBMP, iVMP, and nVMP) were specific for BuChE. All of these esters exhibited AChE-mediated cleavage rates well below the accurate detection limit of 0.004 ΔA/min/U/L, even at enzyme concentration of 100 U/mL. For comparison, AChE-mediated cleavage data for only the n-butyrate esters, BCh and nBMP, are included in Table 2. These data are in agreement with structure-activity relationship studies involving the choline esters (Whittaker, 1951) and with previous work on the piperidinyl esters (Snyder et al., 2000). The straight chain n-valerate ester (nVMP) reacted more rapidly with BuChE than nBMP (Table 3). AMP was cleaved by BuChE at one third the rate of nBMP, similar to the relative cleavage rates of ACh and BCh (BuChE cleavage of BCh/ACh = 3.7:1). Ester branching, either α (iBMP, 2MBMP) or β (iVMP) to the carbonyl, dramatically reduced the rate of cleavage by both ChE enzymes. The cleavage of these branched esters by either enzyme at 20 U/mL was below accurate detection limits, so assays were repeated at 100 U/mL to provide data for comparison.
Based on these in vitro data, nBMP and nVMP were selected for radiolabeling with 11C and further evaluation in animal models as radiotracers for BuChE enzymatic activity. The poor BuChE substrate 2MBMP was also selected for 11C-labeling as a negative control to assess the predictive value of the spectrophotometric assay as an indicator of radiotracer behavior in vivo.
In vitro ester cleavage rate: whole blood
Simplified in vivo pharmacokinetic analyses, which depend upon complete radiotracer metabolism to 1-[11C]methylpiperidinol and trapping of the radioactivity in either brain or periphery, have been developed for [11C]PMP in both human (Koeppe et al., 1999) and rodent (Kilbourn et al., 1999) models. Because of the short half-life of 11C (t1/2 = 20.3 minutes), these simplified pharmacokinetic analyses require that metabolic trapping be complete by 80 to 90 minutes postinjection of the radiotracer. Because ChE enzymes are present in both blood and brain tissues, it is essential to assay the metabolism of candidate radiotracers before interpretation of in vivo data. It has been demonstrated chromato-graphically for both [11C]AMP and [11C]PMP that the only radioactive metabolite formed in mouse brain tissue in vivo is 1-[11C]methylpiperidinol (Irie et al., 1994; Kilbourn et al., 1996). Similarly, assay of blood samples from either mice (Kilbourn et al., 1996) or humans (Namba et al., 1999) has shown no further peripheral metabolism of these radiotracers or of [11C]iBMP (Snyder, unpublished data), other than that mediated by blood ChE. Furthermore, the authors have demonstrated that the fraction of total radioactivity attributed to intact [11C]PMP at 10 and 30 minutes postinjection of radiotracer is less in brain tissue than in blood. Assuming in vivo metabolism is at least as rapid as in vitro (room temperature), it should be sufficient to perform metabolite analysis for candidate radiotracers in whole blood in vitro at room temperature.
The 11C-labeled analogs of the BuChE specific substrates nBMP, 2MeBMP, and nVMP were incubated in whole mouse blood, in vitro at room temperature, and hydrolysis rates were determined by thin-layer chromatography. Blood samples containing [11C]PMP were also analyzed for purposes of comparison with previous in vivo data (Kilbourn et al., 1996). Results for these radiotracers are shown in Fig. 2. As expected, [11C]PMP metabolism in vitro at room temperature was slower than in vivo. Approximately 12% of the radioactivity at 10 minutes was authentic radiotracer as compared with only 4% in vivo (Kilbourn et al., 1996). However, metabolism both in vitro and in vivo was complete by 30 minutes. For the BuChE specific substrates, the rank order of hydrolysis was consistent with the hydrolytic rates determined using the purified enzymes, with the branched ester [11C]2MeBMP hydrolyzed much more slowly than the straight chain esters. The BuChE-mediated cleavage rates determined for PMP and nBMP using purified enzyme were similar, but the AChE-mediated cleavage of PMP was also quite rapid. Thus, in whole blood, [11C]PMP was hydrolyzed more rapidly than [11C]nBMP.
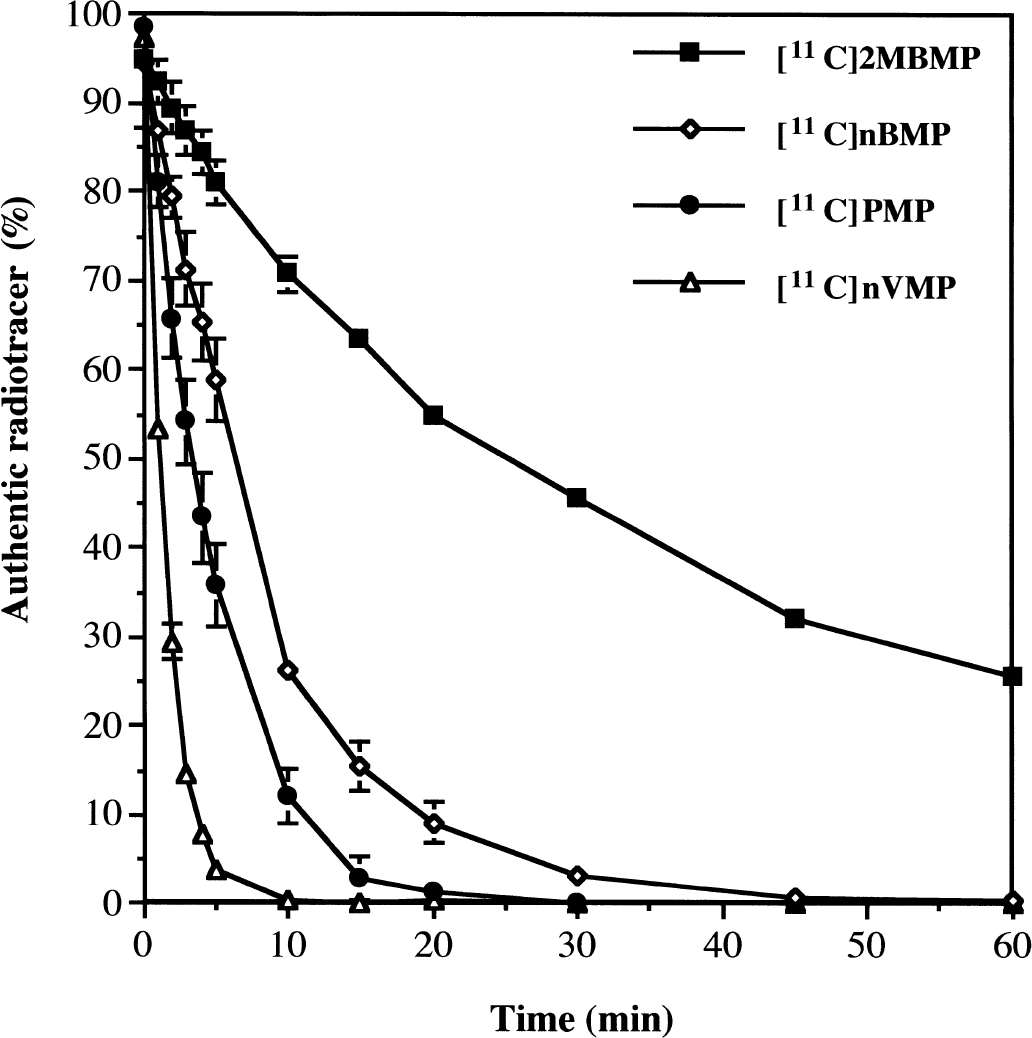
Metabolism of radiotracers in mouse whole blood. Data are expressed as the percentage of authentic radiotracer remaining (means ± SD) at each time point. Curves for [11C]2MBMP and [11C]nVMP represent 1 chromatographic assay using blood from 3 animals. Curves for [11C]nBMP and [11C]PMP represent mean ± SD for 3 separate assays (n = 3 animals per assay).
[11C]nVMP and [11C]nBMP were completely metabolized in whole blood, at room temperature, within 10 and 30 minutes, respectively. Again, these metabolic rates were in good agreement with the relative cleavage rates in purified enzyme (nVMP:nBMP = 2:1). The actual metabolic rate in the living mouse periphery would be expected to be even more rapid, thus complete trapping of radioactivity should be achieved by 30 minutes postinjection of either radiotracer. These in vitro data indicated that the simplified rodent pharmacokinetic model (Kilbourn et al., 1999) could be used for analyzing mouse biodistribution experiments involving both [11C]nVMP and [11C]nBMP. As metabolism of [11C]2MBMP was still incomplete at 90 minutes postinjection, it would not have been a useful 11C-labeled radiotracer and was not evaluated in vivo.
In vivo biodistribution in mouse brain
Representative time-radioactivity curves for the retention of [11C]nBMP and [11C]nVMP in mouse brain are shown in Figure 3. These BuChE specific substrates exhibited high initial brain uptake at 1 minute postinjection, comparable with [11C]PMP (∼10% injected dose/g in cortex) (Kilbourn et al., 1999). The washout rates of both [11C]nBMP and [11C]nVMP were very rapid with <2% injected dose/g trapped in tissues at 30 minutes postinjection of radiotracer, as compared with 4% injected dose/g cortex for [11C]PMP. After 30 minutes, the amount of radioactivity remained constant in all brain regions, indicating complete metabolism of radiotracer in both the central nervous system and periphery and irreversible trapping of the radiolabeled metabolite in brain tissue. Both initial uptake and retention of radioactivity were significantly greater for [11C]nVMP than for [11C]nBMP, which is consistent with the more rapid cleavage of the former by BuChE measured in vitro.
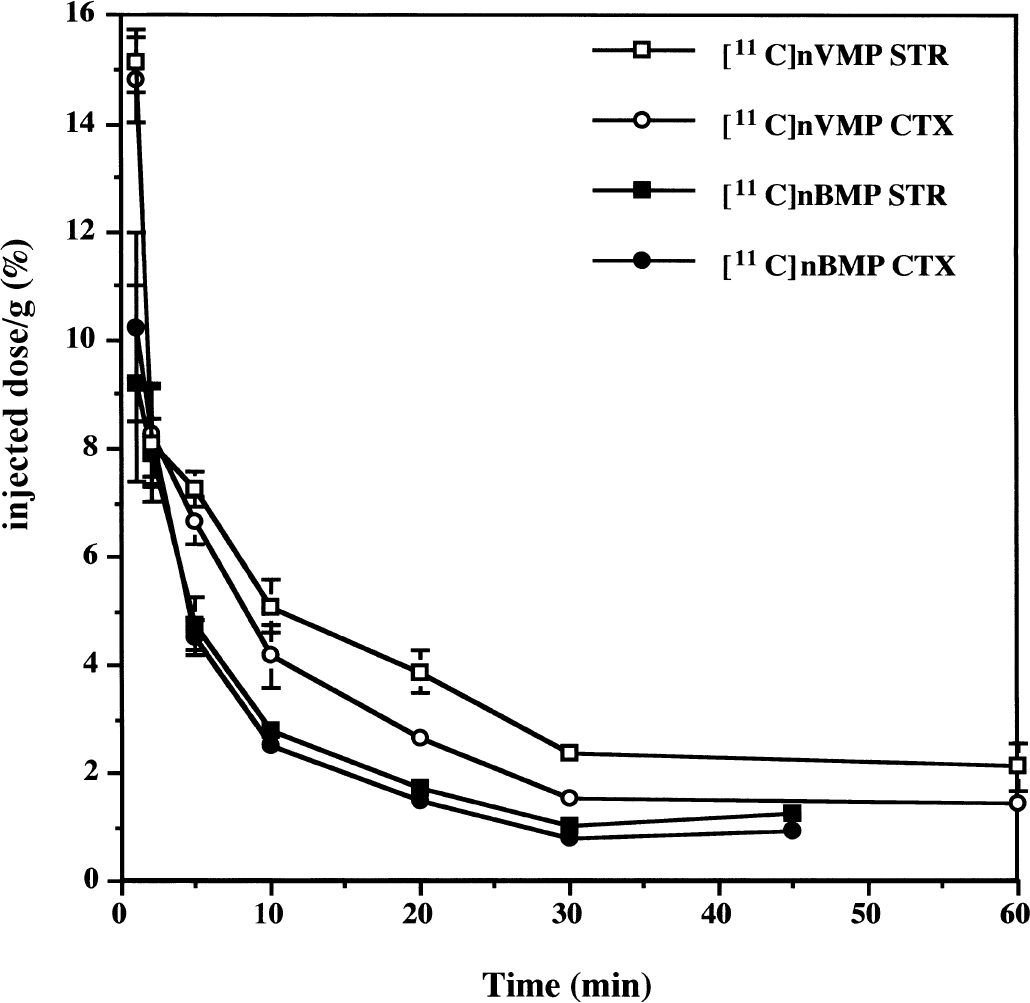
Time-activity curves for [11C]nVMP and [11C]nBMP in mouse brain. Female CD-1 mice were injected with 150 to 250 μCi of radiotracer then killed at various times postinjection. Data are expressed as the percent injected radioactivity dose per gram of tissue in striatum (STR) and cortex (CTX) at each time point. Each point is the mean ± SD for n ≥ 4 animals.
The retention fraction in each brain region was calculated as the ratio of radioactivity retained at 30 minutes to the initial uptake of radioactivity at 1 minute postinjection of radiotracer. As reported previously (Kilbourn et al., 1999), these retention fractions are an index of the ChE activity in each brain region, provided metabolism and trapping of the radiotracer is complete by 30 minutes postinjection. Also, the use of retention fractions, rather than absolute radioactivity retention values, lessens the effects of blood flow and interexperiment variation. The results of these calculations for [11C]nBMP and [11C]nVMP are summarized in Table 4, with [11C]PMP included here for comparison purposes.
In vivo regional distribution in mouse brain
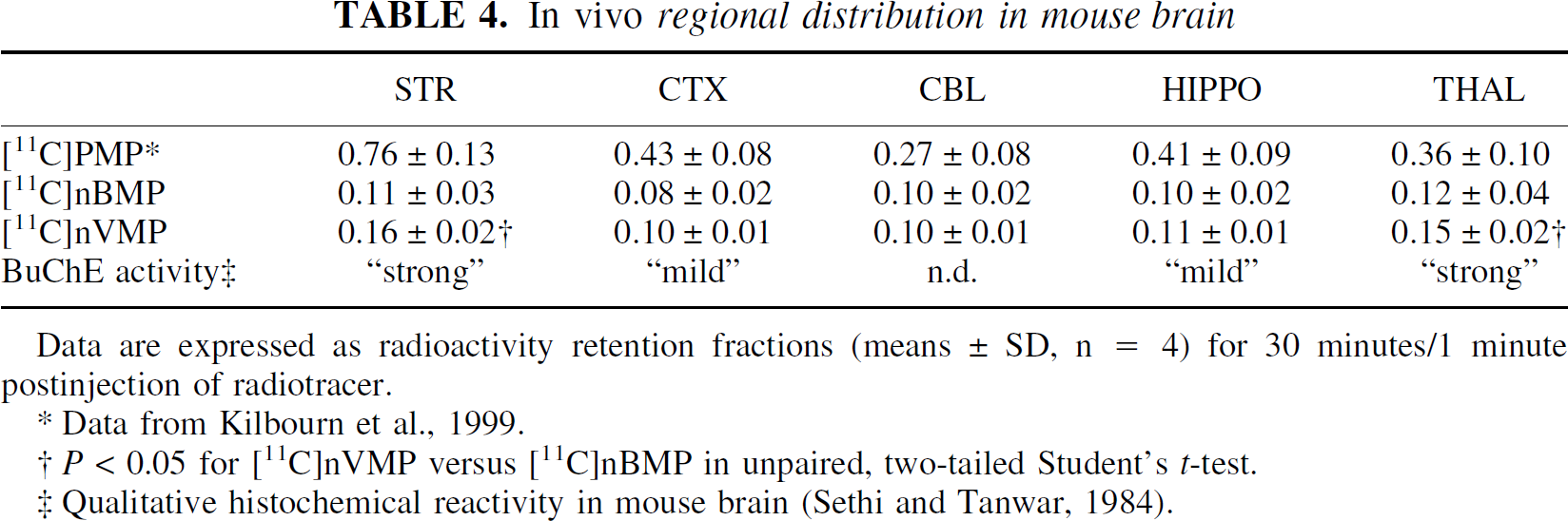
Data are expressed as radioactivity retention fractions (means ± SD, n = 4) for 30 minutes/1 minute postinjection of radiotracer.
Data from Kilbourn et al., 1999.
P < 0.05 for [11C]nVMP versus [11C]nBMP in unpaired, two-tailed Student's t-test.
Qualitative histochemical reactivity in mouse brain (Sethi and Tanwar, 1984).
Retention fractions in all brain regions were much less for the BuChE specific radiotracers than for [11C]PMP. Retention of [11C]nVMP in striatum and thalamus was significantly greater (P < 0.05) than [11C]nBMP. Retention in all other brain regions was similar for the two radiotracers. The regional distributions of both [11C]nBMP and [11C]nVMP are consistent with a detailed, but qualitative, BuChE distribution in mouse brain (Sethi and Tanwar, 1984). This histochemical investigation reports “strong” BuChE reactivity in caudate nucleus, putamen, and various thalamic nuclei. A “mild” BuChE reactivity was seen in neocortex, hippocampus, and hypothalamus.
Despite these differences in regional distribution, no definitive distinction in radiotracer efficacy could be made, based solely on rodent data, between [11C]nBMP and [11C]nVMP. Therefore, both radiotracers were evaluated in PET imaging experiments in primates as a more accurate model of the potential of these radiotracers for in vivo imaging in humans.
In vivo imaging in primates
Postron emission tomography imaging experiments using [11C]AMP and [11C]PMP in primates have been performed previously (Kilbourn et al., 1996). Using a similar protocol, a female pigtail monkey (M. nemistrina) was injected with [11C]nBMP or [11C]nVMP and imaged for 60 minutes. Time-activity curves for striatum and cortex are shown in Fig. 4, with [11C]PMP data again included for comparison. Data are from a single animal scan for each radiotracer but using the same animal for all three scans and data are normalized to the injected dose of radioactivity. In contrast to the rodent data, initial uptake in primate brain was much less for both [11C]nBMP and [11C]nVMP relative to [11C]PMP. This difference may reflect a species difference in the blood BuChE concentration, resulting in higher BuChE-mediated peripheral metabolism of radiotracer in primates.
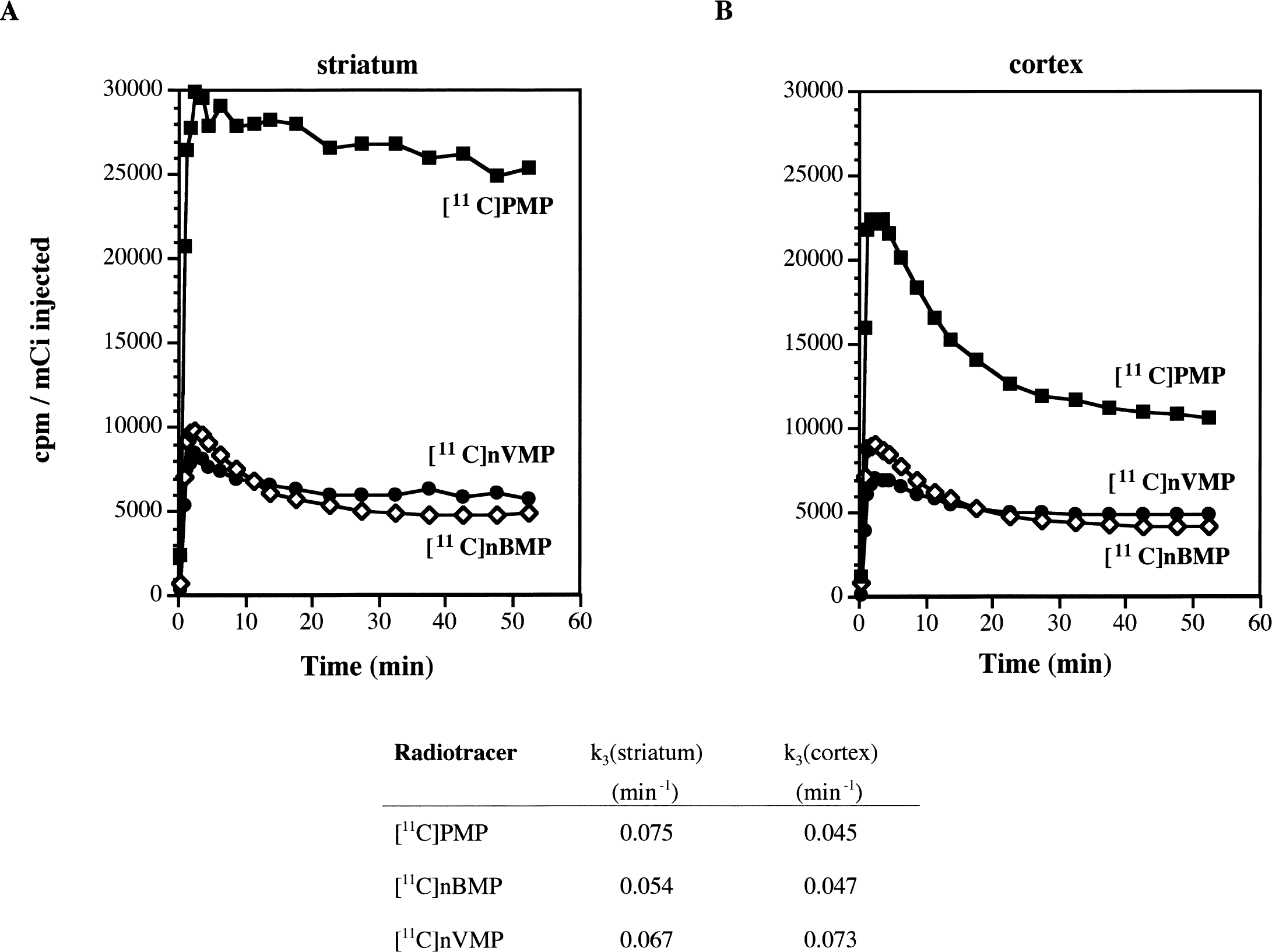
Positron emission tomography (PET) imaging data for [11C]PMP, [11C]nBMP, and [11C]nVMP in primates. Data represent time-activity curves and apparent enzymatic rate constants (k3) (Koeppe et al., 1999) for regions of interest drawn over
Initial brain uptake of [11C]nVMP was significantly less than [11C]nBMP, yet both radiotracers exhibited similar amounts of radioactivity trapped at later time points. Again consistent with the in vitro data, the lower initial uptake of [11C]nVMP is likely a result of rapid peripheral hydrolysis. Similarly, 70% to 80% of the [11C]nVMP entering the brain is rapidly metabolized and trapped in tissue versus approximately 50% trapping of radioactivity for [11C]nBMP.
The apparent hydrolysis rate constants k3 (Koeppe et al., 1999), calculated from these PET data are included in Fig. 3. This k3 value reflects both the actual rate of enzymatic hydrolysis (khyd) and the local concentration of the enzyme ([ChE]). For the BuChE specific substrates, k3 is proportional to khyd[BuChE] in each region. Again consistent with the in vitro cleavage rates, the k3 values for [11C]nVMP in striatum and cortex were larger than those for [11C]nBMP.
DISCUSSION
The majority of investigators performing spectrophotometric assays on the cholinesterases are doing so to quantify levels of enzyme in tissue samples (Rappaport et al., 1959; Augustinsson et al., 1978; Finkelstein et al., 1988; Hammond and Brimijoin, 1988; Huff et al., 1989) or to identify new enzyme inhibitors (Pang et al., 1996; Taylor, 1998). Little research has been reported in the recent literature to identify novel substrates for ChE, likely owing to the lack of pharmacologic use for such compounds. Not surprising then, most of the cholinesterase assays reported in the literature depend on standard substrates, such as the thiocholine esters (Augustinsson et al., 1978; Vellom et al., 1993). Few non-radiometric options are available for screening multiple substrates. The authors have adapted such a substrate-independent kinetic assay from a standard clinical toxicology screen, originally designed to determine blood cholinesterase levels in poisoning victims (Rappaport et al., 1959). By means of this spectrophotometric assay, the authors identified two new radiotracer candidates, [11C]nBMP and [11C]nVMP, for further in vitro and in vivo examination and avoided the radiolabeling and evaluation of compounds that did not meet kinetic or selectivity requirements.
The spectrophotometric assay was performed at saturating substrate concentrations (50 mg/mL), whereas the in vitro metabolism and in vivo distribution studies were performed at trace substrate concentrations. Therefore, no comparison of absolute cleavage rates among these experiments is possible. However, relative comparisons between substrates are valid, and in fact there is good correlation between the spectrophotometric data and both in vitro metabolism rates and k3 values measured from PET studies. In terms of Michaelis–Menten kinetics, the cleavage rates measured in the enzyme assay are proportional to the Vmax for this reaction. The absolute radiotracer cleavage rates (V) for the in vivo experiments would be dependent on the substrate concentration ([S]) which is not known. However at these low substrate concentrations, the relative rate differences between substrates (for example, nBMP vs. nVMP) are primarily dependent on the Km for each substrate; that is, at tracer concentrations, BuChE-mediated cleavage of nVMP will always be more rapid than cleavage of nBMP. A radiometric assay for ChE-mediated cleavage of the methylpiperidinyl esters at near tracer concentration has been reported (Irie et al., 1994). As discussed previously, the results of the authors' spectrophotometric assay are in excellent agreement with the radiometric data.
Cleavage rates for the piperidinyl esters described here, assayed at 37°C, have been reported previously in abstract form (Snyder et al., 2000). However, no details of the assay method were provided and no attempt was made to rigorously assess any effect of enzyme concentration on specific rate comparisons. A temperature of 25°C was chosen for this assay, rather than 37°C as previously reported, to minimize nonlinearity in assays of the more rapidly cleaved substrates.
This nonlinear absorbance change may have resulted from competing buffering effects of the assay mixture or depletion of substrate during the assay. Although the reactivity of both enzymes is pH-dependent, a change in pH during the assay does not adequately explain this observation, because the absorbance change measured during a typical assay resulted from a <0.2 pH unit decrease (that is, pH 7.8 to 7.6). Likewise, the assay is run at pH 7.8 and the enzymatic rates for AChE and BuChE remain relatively constant between pH 8.0 and 7.5 (Baum and Ward, 1971; Ashihara et al., 1983).
Based on the temperature dependence of both enzymes (King and Morgan, 1970; Spinedi et al., 1988), it was anticipated that lowering the assay temperature would reduce the overall enzymatic rates and provide more accurate data. In fact, assays of ACh and BCh at 25°C were more reproducible and the cleavage rates observed were much greater than previously reported (Snyder et al., 2000). However, there were some discrepancies in the enzymatic rates measured at these two different temperatures. Most notably, the BuChE-mediated cleavage rate of PMP at 25°C was somewhat faster than that of nBMP. In the previous experiment, the rate of substrate cleavage by BuChE was directly correlated with ester chain length (nVMP > nBMP > PMP > AMP). The explanation for this discrepancy is unclear.
Also, the relative cleavage rates for nBMP and nVMP (Table 3) measured at 25°C more accurately reflected the large in vitro metabolic rate difference for these substrates in whole blood (Fig. 1). At 25°C, nVMP exhibited an in vitro BuChE cleavage rate approximately twice that of nBMP, whereas at 37°C this ratio was approximately 1.2:1 (Snyder et al., 2000). This compression of the relative cleavage rates by both ChE enzymes was a general characteristic of the previously reported spectrophotometry data. It is unclear whether the compression of relative in vitro cleavage rates at 37°C is an actual temperature-dependent change in enzyme kinetics or merely reflects an inherent inaccuracy in the rates measured at the higher temperature. However, the data collected at 25°C were also more predictive of the substantial in vivo difference observed between [11C]nBMP and [11C]nVMP in both rodents and primates.
Some apparent differences in radiotracer localization were also observed in vivo between rodent and primate data. These likely result from the distribution of ChE in both the central nervous system and in the periphery and significant species variations. Radiotracers based on ChE substrates take advantage of enzymatic cleavage as a mechanism of localization. The [11C]methylpiperidinyl esters freely diffuse across the blood–brain barrier, but once cleaved, the more hydrophilic N-[11C]methylpiperidinol is trapped in tissue. Thus, the level and rate of radioactivity trapping in a given region provides an index of ChE activity in that region (Irie et al., 1994). In addition to neuronal localization, both AChE and BuChE are present at nanomolar concentrations in blood (Brimijoin and Hammond, 1988). Studies using [14C]methylpiperidinol (Irie et al., 1994) have shown that the labeled metabolite formed by this peripheral ChE activity is unable to enter the brain. Thus, the high blood ChE activity has a large influence on the biodistribution and pharmacokinetics of radiotracers for these two enzymes. This is particularly important for development of radiotracers for BuChE because the whole blood BuChE activity in humans is approximately three times that of AChE (Brimijoin and Hammond, 1988).
In addition, large species differences in the concentrations of AChE and BuChE in both blood and brain are known to exist (Sethi and Tanwar, 1984; Carmona et al., 1996). Although the AChE and BuChE concentrations in human cortex are similar (Atack et al., 1986; Brimijoin and Hammond, 1988), the AChE: BuChE ratio is approximately 10:1 in most brain regions in the adult rat (Lassiter et al., 1998). The authors observed significant variation in the metabolism of [11C]PMP between mice (Kilbourn et al., 1996) and humans (Koeppe et al., 1999). Likewise, in vitro whole blood metabolism of [11C]nBMP is nearly 10 times faster for mice than for rats (Snyder, unpublished data). The relative concentrations of AChE and BuChE in the mouse brain are not well characterized. However, the lower cortical retention fractions in mice for [11C]nBMP and [11C]nVMP, as compared with[11C]PMP (Table 4), may reflect a species-specific paucity of BuChE, relative to AChE, in mouse brain, similar to that observed in rat. This difference may also reflect a contribution from both the AChE and BuChE components of [11C]PMP cleavage to the overall rate of metabolic trapping, whereas [11C]nBMP and [11C]nVMP are BuChE specific.
Despite the lower retention of [11C]nBMP and [11C]nVMP versus [11C]PMP, the initial uptake for all three radiotracers in mouse brain was ≥10% injected dose/g (Fig. 2) (Kilbourn et al., 1996). In contrast, the maximal uptake values for [11C]nBMP and [11C]nVMP in primate cortex (Fig. 4) were 40% and 30% that of [11C]PMP, respectively. Coupled with the spectrophotometry and metabolism results, these data suggest a greater peripheral BuChE concentration in primates than in mice. This also would account for the initial uptake differences between [11C]nBMP and [11C]nVMP in primate brain (Fig. 4) that were opposite of those in mice (Fig. 3). At high peripheral BuChE concentrations, the more rapid cleavage of nVMP versus nBMP (Table 3) would lead to lower concentrations of unmetabolized [11C]nVMP in blood available for uptake into brain tissues.
This difference in BuChE-mediated cleavage rates between [11C]nBMP and [11C]nVMP was also observed in the pharmacokinetics of radioactivity retention in mouse and primate brain. Significantly higher retention fractions were observed (Table 4) for [11C]nVMP in mouse striatum and thalamus. In primates, striatal and cortical k3 values for [11C]nVMP (Fig. 4) were 25% and 55% greater, respectively, than the corresponding values for [11C]nBMP. As discussed previously, measurement of both retention fractions in rodents and estimates of k3 in primates depend on complete metabolism of the radiotracer in vivo to form exclusively 1-[11C]methyl-4-piperidinol. Based on the in vitro metabolism data and on previous human (Namba et al., 1999) and rodent (Kilbourn et al., 1996) in vivo evidence using [11C]PMP, both [11C]nBMP and [11C]nVMP should be completely converted to 1-[11C]methyl-4-piperidinol in less than 30 minutes in vivo. However, this assumption has not been tested directly.
Reactivity of both nBMP and nVMP with BuChE in vitro, and strong agreement between in vitro predictions and in vivo observations indicate a mechanism of localization for these esters analogous to that of [11C]PMP—that is, specific cleavage of [11C]nBMP or [11C]nVMP by BuChE, followed by tissue trapping of the labeled cleavage product. Rigorous validation of this mechanism using enzyme inhibition studies is complicated by the presence of BuChE in brain and periphery and by an inability to completely inactivate the enzyme in vivo (Kilbourn et al., 1999; Kuhl et al., 2000). It is possible to generate inhibitor dose-response data in mice using retention fractions (Kilbourn et al., 1999), however such experiments were beyond the scope of these initial studies with [11C]nBMP or [11C]nVMP.
The in vivo pharmacokinetic distinction between [11C]nBMP and [11C]nVMP ultimately led to the selection of [11C]nBMP for evaluation as a radiotracer for the functional imaging of BuChE in humans with PET. In striatum and cortex, the primate k3 values for [11C]nVMP were similar to that of [11C]PMP in striatum (Fig. 4), yet were still less than the corresponding values for [11C]AMP (k3(str) = 0.117 and k3(ctx) = 0.085), a radiotracer known to be delivery-dependent in striatum (Koeppe et al., 1999; Shinotoh et al., 2000)—that is, the high concentration of AChE in striatum leads to a very rapid cleavage of [11C]AMP such that the rate-limiting step for radioactivity trapping becomes the rate of diffusion across the blood–brain barrier. In human cortex, [11C]AMP approaches the upper limit of cleavage rate to provide accurate measures of cortical AChE activity (Koeppe et al., 1999; Shinotoh et al., 2000). Similarly, the more rapid hydrolysis of [11C]nVMP by BuChE may lead to difficulties with delivery dependence, particularly in light of the large increases in BuChE concentration observed postmortem in patients with AD (Perry et al., 1978; Mesulam and Geula, 1994).
This possible limitation, coupled with the lower overall uptake of radioactivity into primate brain for [11C]nVMP, indicates that [11C]nBMP may be the better choice for measuring BuChE activity in human subjects. In both mice and primates, [11C]nBMP exhibits adequate extraction into brain and a regional retention of radioactivity consistent with the reported distribution of BuChE histochemical reactivity (Sethi and Tanwar, 1984; Atack et al., 1986). The apparent enzymatic rate constant k3) in primate brain for [11C]nBMP is in a range in which it can be estimated with good precision (Koeppe et al., 1999) and should be sensitive to both increases and decreases in BuChE in disease states. Through use of this new radiotracer in human volunteers, questions about the role of BuChE in the etiology and progression of neurodegenerative diseases such as AD can now be investigated in vivo.
Footnotes
Acknowledgments:
The authors thank Kyle Kuszpit and Leslie Doherty for their assistance with the animal studies and the cyclotron staff at the University of Michigan PET Facility for radionuclide production.