
Abstract
Insulin-like growth factor (IGF-1) is induced in damaged brain tissue after hypoxia—ischemia, and exogenous administration of IGF-1 shortly after injury has been shown to be neuroprotective. However, it is unknown whether treatment with IGF-1 delayed by more than a few hours after injury may be protective. Hypothermia after brain injury has been reported to delay the development of ischemic neuronal death. The authors therefore hypothesize that a reduction in the environmental temperature during recovery from hypoxia—ischemia could prolong the window of opportunity for IGF-1 treatment. Unilateral brain damage was induced in adult rats using a modified Levine model of right carotid artery ligation followed by brief hypoxia (6% O2 for 10 minutes). The rats were maintained in either a warm (31°C) or cool (23°C) environment for the first 2 hours after hypoxia. All rats were subsequently transferred to the 23°C environment until the end of the experiment. A single dose of IGF-1 (50 μg) or its vehicle was given intracerebroventricularly at either 2 or 6 hours after hypoxia. Histologic outcome in the lateral cortex was quantified 5 days after hypoxia. Finally, cortical temperature was recorded from 1 hour before and 2 hours after hypoxia in separate groups of rats exposed to the “warm” and “cool” protocols. In rats exposed to the warm recovery environment, IGF-1 reduced cortical damage (P < 0.05) when given 2 hours but not 6 hours after insult. In contrast, with early recovery in the cool environment, a significant protective effect of IGF-1 in the lateral cortex (P < 0.05) was found with administration 6 hours after insult. In conclusion, a reduction in cerebral temperature during the early recovery phase after severe hypoxia—ischemia did not significantly reduce the severity of injury after 5 days' recovery; however, it markedly shifted and extended the window of opportunity for delayed treatment with IGF-1.
Insulin-like growth factor (IGF)-1 is a broadly neurotrophic factor widely distributed in the CNS (Baskin et al., 1988). Both IGF-1 mRNA and protein are potently induced in damaged regions after brain injury (Gluckman et al., 1998; Gluckman et al., 1992). The time course of endogenous IGF-1 induction varies from 24 hours to 3 weeks, depending on the severity of injury. This delayed induction after brain injury may either improve recovery or act to limit the extent of neuronal loss by providing trophic support to a stressed system (Iihara et al., 1994; Klempt et al., 1992). The neuroprotective effects of exogenous IGF-1 given soon after brain injury have been demonstrated in various experimental animal models (Johnston et al., 1996; Guan et al., 1993; Gluckman et al., 1992). However, to be clinically useful, a broad window of therapeutic opportunity is desirable.
Some neurons die many hours or even days after brain injuries, with more rapid evolution associated with more severe insults (Beilharz et al., 1995; Li et al., 1995). This delayed evolution of neuronal death provides a potential window of opportunity for treatment. Little information is available on the factors other than ultimate cell death that determine the practical limits of this treatment window. It is more likely, for example, that neurotrophic factors may be protective if applied during the initial “latent” phase of programed cell death than during the active phase, although this still precedes DNA fragmentation and cell death (Samejima et al., 1998).
Hypoxia—ischemia is commonly associated with a decline in brain temperature during and for a short period after an insult, unless it is actively prevented by periinsult warming (Minamisawa et al., 1990). The effect of such spontaneous mild hypothermia limited to the recovery period is unclear (Gunn, Gunn, 1998). Several studies in adult and newborn animals report that short periods of active postinsult cerebral cooling can delay neuronal death (Trescher et al., 1997; Coimbra et al., 1996; Dietrich et al., 1993;. Dietrich et al., 1995). Consistent with this, moderate to deep (30°C) hypothermia for 3 hours after ischemia in the rat markedly increased the effect of delayed treatment with a noncompetitive N-methyl-
In the current study, we examine the hypothesis that injury-induced postischemic hypothermia would broaden the period when IGF-1 could be administered as a neuroprotective agent.
METHODS
Animal preparation
The study was approved by the Animal Ethics Committee of the University of Auckland. All efforts were made to minimize animal suffering and to reduce the number of animals used. Surgical procedures were carried out under halothane (3%) anesthesia. A block experimental design was used for each experimental group as summarized in Table 1. Animals in each experimental block of six rats underwent hypoxia and subsequent vehicle or IGF-1 treatment simultaneously. Adult rats (Wistar, male, 280 to 310 g) were used in these studies. The modified Levine preparation and experimental procedures have been previously described (Guan et al., 1993). Briefly, a lateral ventricular cannula was implanted, and then hypoxic—ischemic (HI) injury was induced by unilateral carotid artery ligation followed by inhalational hypoxia.
Experimental groups
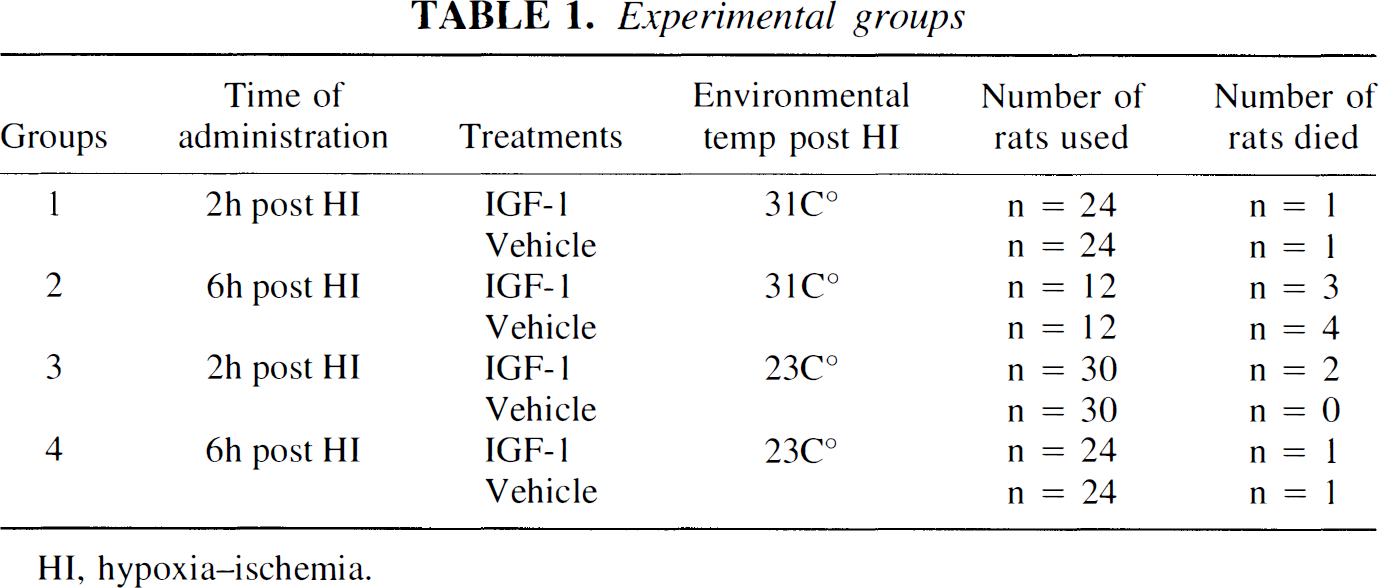
HI, hypoxia—ischemia.
The detailed procedures were as follows. A guide cannula was stereotaxically placed on the top of the dura 1.5 mm to the right of the midline and anteroposteriorly (A-P) 7.5 mm from the interaural zero plane. One day later, the right carotid artery was double-ligated. After 1-hour recovery from anesthesia, the rats were placed in an incubator with controlled humidity (90% ± 5%) and temperature (31° ± 0.5°C) for another hour, then exposed to 10 minutes of hypoxia (6% ± 0.2% oxygen). The rats recovered in either a “cool” or a “warm” environment. The cool recovery environment was a holding room with controlled temperature (23° ± 1°C) and humidity (50% ± 5%), where they were kept until the termination of experiments (Table 1). For the warm recovery groups, rats were kept in the incubator for 2 hours after hypoxia, then transferred into the same holding room and held until the termination of experiments (Table 1). Rats within each experimental group were treated simultaneously with either IGF-1 (50 μg) or its vehicle given intracerebroventricularly at 2 or 6 hours after the hypoxia under anesthesia with 3% halothane. A total volume of 25 μL was infused intracerebroventricularly over 12.5 minutes using a microinfusion pump (Guan et al., 1993). The rats were killed with an overdose of sodium pentobarbital 5 days after hypoxia for histologic analysis. Rats that died before the termination of experiments were rejected from histologic analysis and were treated statistically as missing data.
Drug preparation
The IGF-1 (8 mg/mL, Myotrophin, Chiron Ltd., Emeryville, CA, U.S.A.) was stored in acetate buffer (pH 4.0) and neutralized with a half-volume of 8.4% NaHCO3 and then diluted three times with 0.1% bovine serum album/0.1 mol/L phosphate-buffered saline (pH 7.3) immediately before intracerebroventricular infusion.
Histologic preparation and analysis. The histologic procedures have been previously described in detail (Guan et al., 1996; Guan et al., 1993). Briefly, the brains were perfused transcardially with normal saline followed by phosphate-buffered formalin (10%, pH 7.3). The brains were kept in the same fixative for 2 days before being processed using a standard paraffin procedure. Coronal (5-μm) sections were cut and mounted on glass slides and stained with thionine and acid fuchsin. The percentage of damage in the lateral cortex was assessed using three levels: A, middle level striatum (A-P 6 mm); B, middle level hippocampus (A-P 4.5 mm); and C, level showing both the ventral and dorsal horns of the hippocampus (A-P 4.2 mm, Fig. 1). The lateral cortex at each level was divided into three equal regions, and cortical tissue damage was assessed using the following scoring system; 0, no damage; 1, less than 5% tissue damaged; 2, less than 50% tissue damaged; 3, more than 50% tissue damaged; and 4, more than 95% damaged (Lundgren et al., 1992). The tissue damage scores then were transformed into the percentage of tissue damage (Full score 36 = 100%). Cortical tissue with selective neuronal death, cellular reaction, or pan-necrosis was considered to be damaged (Markgraf et al., 1993). Dead neurons were identified as those with acidophilic (red) cytoplasm and contracted nuclei (Auer et al., 1985; Brown, Brierley, 1972).
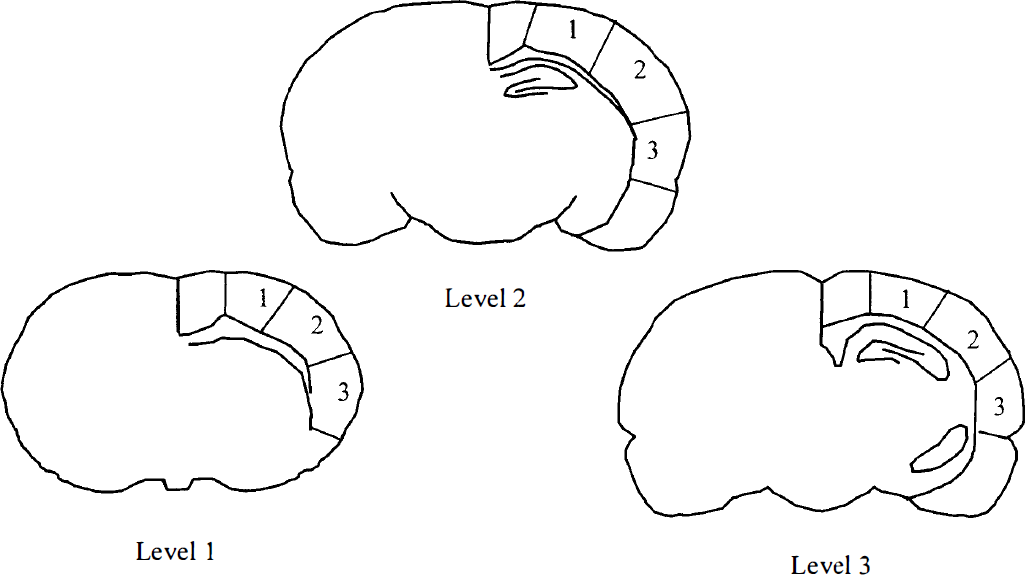
Diagram showing the three coronal sections used to assess cortical tissue damage. The lateral cortex ipsilateral to the hypoxic—ischemic (HI) injury was divided into three regions, and the percentage of tissue damage in each region was scored as described in Methods.
Brain and core temperature recording and analysis. A group of eight rats was used for brain temperature recordings as described previously (Sirimanne et al., 1996). Briefly, a brain temperature probe was implanted on the surface of the cortex approximately A-P 3 to 3.5 mm and 1.7 mm on the right side, 1 day before HI injury. The brain temperature was recorded for 1 hour before hypoxia for the baseline reading and then for a further 2 hours after hypoxia either in the cool (23° ± 1°C and 50% ± 5%) or warm (31° ± 0.5°C and 90% ± 5%) environments. The brain temperature probes were calibrated at room temperature. The brain temperature readings within the incubator (at 31° ± 0.5°C) were corrected for the higher environmental temperature by −0.5°C (Sirimanne et al., 1996). Rats used for brain temperature recordings were evenly distributed between the IGF-1 and the vehicle-treated groups. Brain temperature was analyzed using a customized computer program (LabVIEW, National Instruments, Austin, TX, U.S.A.). The mean temperature from 30-minute intervals was used for data analysis and plotted using the midpoint for each interval. The deep rectal (core) temperature was measured 1 hour before and 0.5, 2, and 4 hours after hypoxia in both warm and cool environments using a digital thermometer.
Statistics. The percentage of tissue damage in the lateral cortex was compared between the IGF-1 and vehicle-treated groups within the time of the treatment by two-way analysis of variance, followed by post hoc testing to define the specific treatment effects. The changes in cortical temperature and core temperature were compared between the warm and cool groups, as well as with baseline recording by analysis of variance, with time treated as a repeated measure, followed by post hoc comparisons (Tukey Test, Sigma Stat, Jandel Scientific Corte Madera, CA, U.S.A.). Histologic and cortical temperature data are presented as mean ± SD.
RESULTS
Cortical temperature and severity of brain damage
The baseline cortical temperature in the holding room (temperature 23° ± 1°C and humidity 50% ± 5%) was 37.4° ± 0.4°C. The cortical temperature recorded in the cool environment (which was the same as in the holding room) during the first 2 hours after HI injury was significantly reduced to 36.1° ± 0.4°C (P < 0.05) compared with baseline. This change was accompanied by a mild reduction in core temperature from 38.7° ± 0.4°C (baseline) to a nadir of 37.9° ± 0.3°C during the period of recovery. The cortical temperature in the warm environment (temperature 31° ± 0.5°C and humidity 90% ± 0.5%) after hypoxia was not statistically increased (39° ± 1.3°C) compared with baseline but was significantly higher than for the rats in the cool recovery environment (P < 0.05, Fig. 2). There was a significant rise in core temperature from 38.6° ± 0.2°C (baseline) to 39.7° ± 0.5°C at 30 minutes after hypoxia (P < 0.05), which then returned to normal (38.6° ± 0.2°C) by 2 hours after hypoxia. The mean core temperature was 38.79° ± 0.2°C in the warm group and 37.93° ± 0.1°C in the cool group.
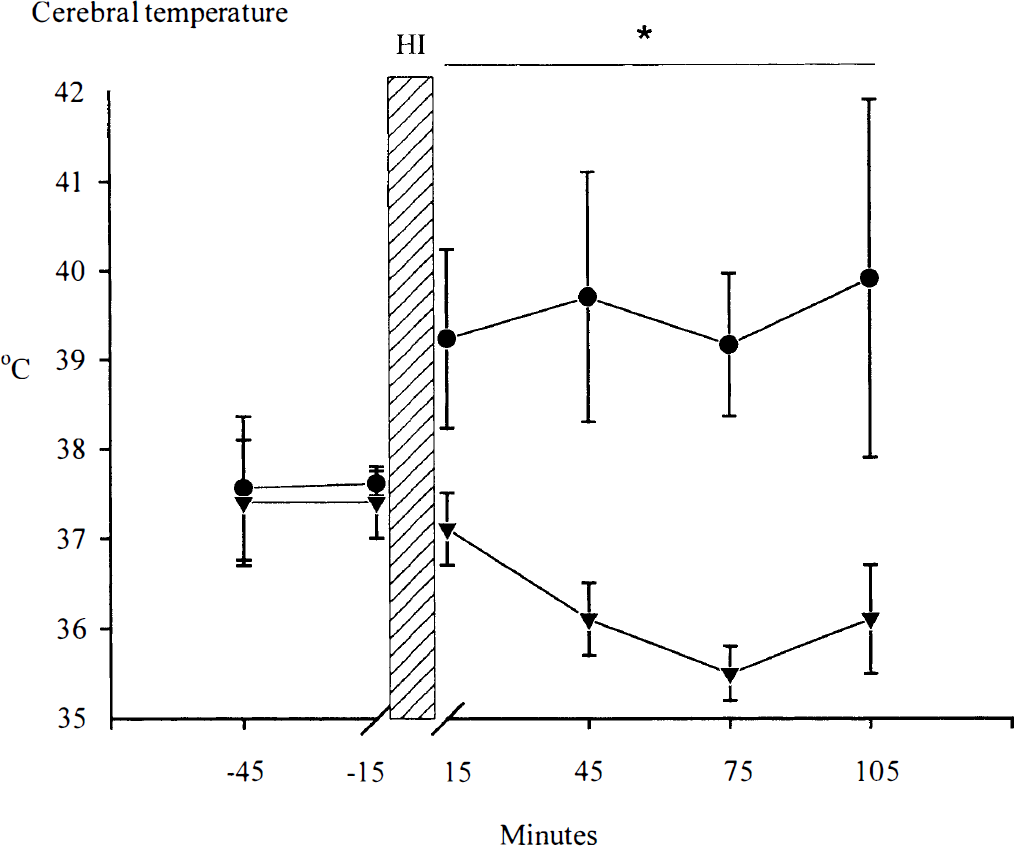
Time sequence of changes in cerebral temperature in rats recovered in the “cool” or “warm” environments. Post-HI cerebral temperature was significantly reduced in the “cool” environment (23°C temperature and 50% humidity) compared with both baseline recordings, and compared with the group recovered in the “warm” environment (31°C temperature and 90% humidity), particularly after 45 minutes after HI. (•) 31 °C (n = 3). (▾) 23°C (n = 4). *P < 0.05; data are presented as mean ± SD.
The percentage of tissue damage in the lateral cortex from vehicle-treated rats, in the main study, was not significantly different between the warm (47.8% ± 43%, n = 36) and cool (38.3% ± 38%, n = 54) groups.
Effects of IGF-1 in rats kept in the warm recovery environment for the first 2 hours
Within the warm group, there was no overall difference between IGF-1 and the vehicle treatments. However, there was a significant interaction between time of administration and treatment effects with IGF-1 or vehicle (P < 0.05). When IGF-1 (50 μg, n = 23) was given 2 hours after HI injury, it significantly reduced the percentage of tissue damage (29.0% ± 43.4%) in the lateral cortex compared with the vehicle-treated group (53.1 % ± 41.0%, n = 23, P < 0.05, Figs. 3 and 4A and 4B), confirming our previous studies (Guan et al., 1996; Guan et al., 1993). However, there was no treatment effect with IGF-1 (58.3% ± 41.7%) administered 6 hours after HI injury compared with the vehicle-treated group (49.5% ± 45.7%, Fig. 3).
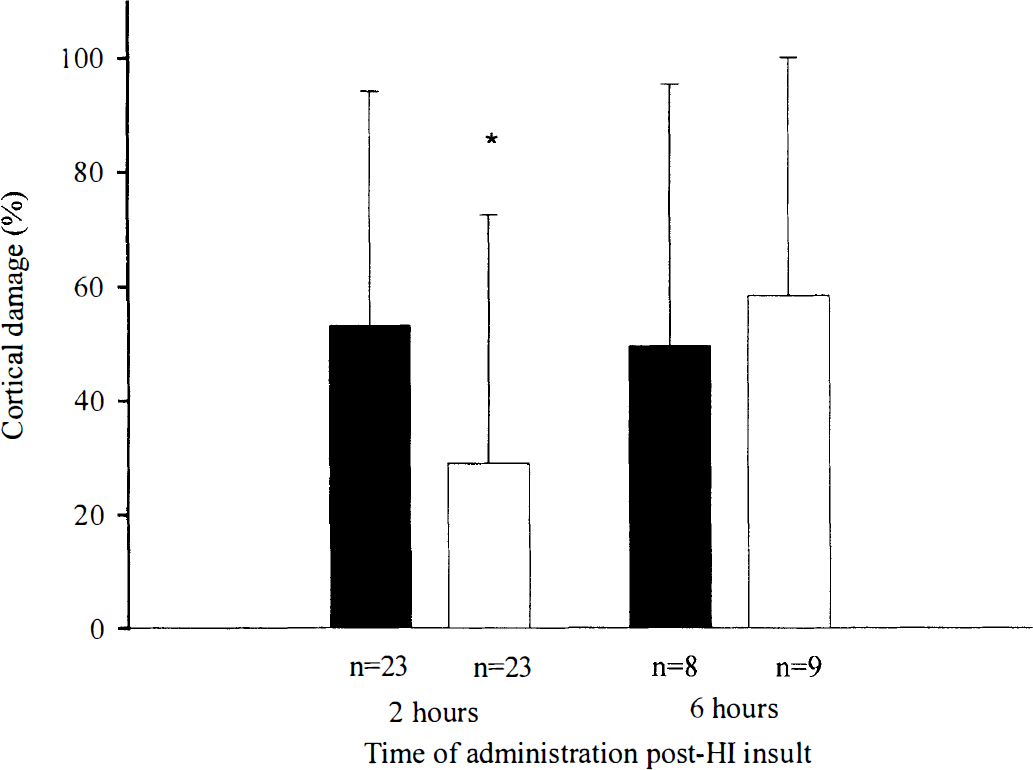
Effects of insulin-like growth factor (IGF-1) treatment on cortical injury in rats exposed to the “warm” recovery environment (31°C) for 2 hours after hypoxia—ischemia. When administered centrally at 2 hours but not 6 hours after hypoxia-ischemia, IGF-1 significantly reduced the percentage of cortical damage. (▪) Vehicle. (□) IGF-1. *P < 0.05 compared with vehicle controls; data are presented as mean ± SD.
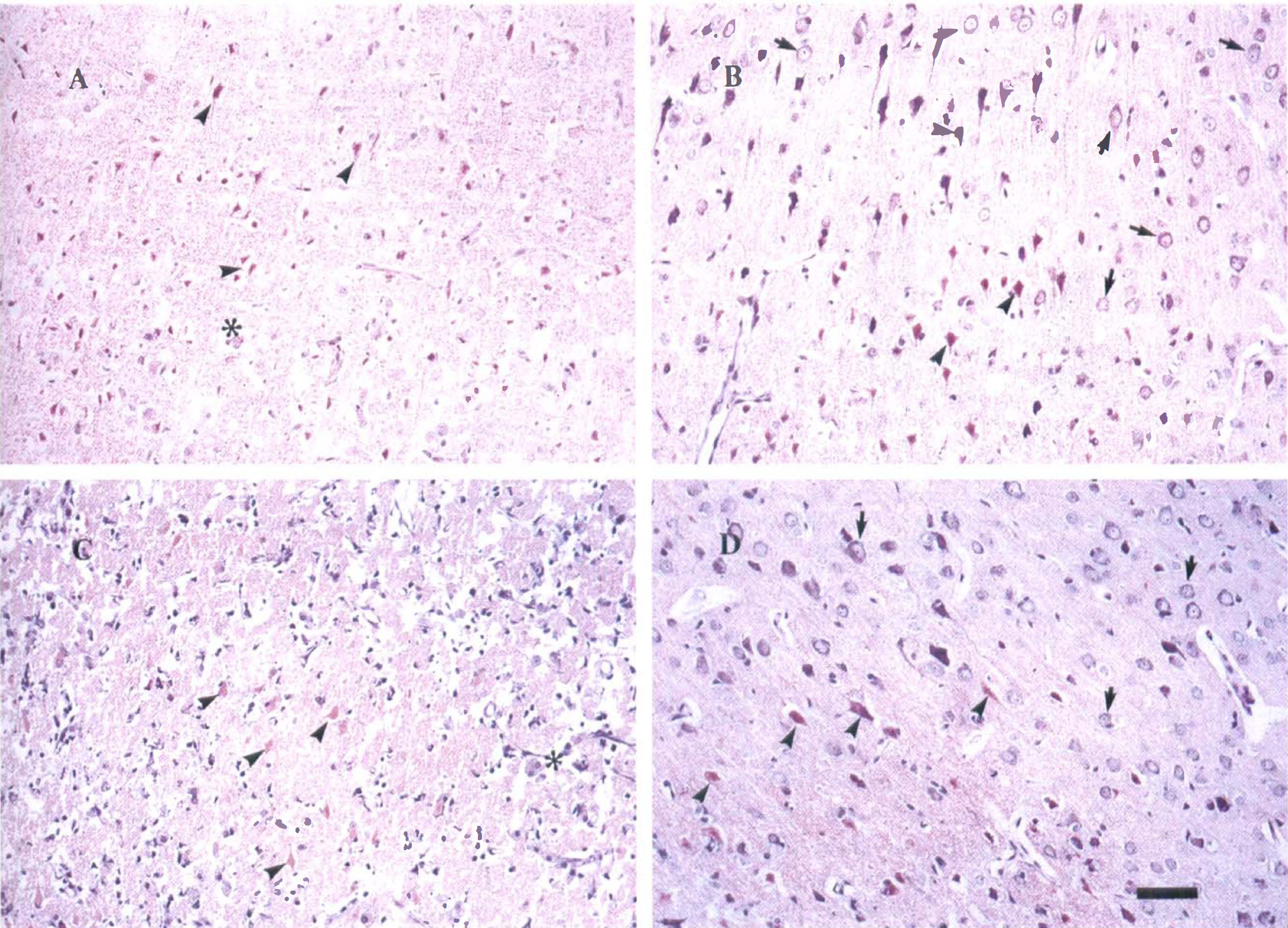
Photomicrographs showing the treatment effect of IGF-1 on neuronal loss. Photographs were taken from the most severely damaged areas. The cortical areas with selective neuronal loss, cellular reaction, and pan-necrosis were considered to be damaged. The tissue infarction (stars), strong cellular reaction, and ischemic neurons (arrowheads) were more pronounced in the vehicle-treated groups from both the “warm” and “cool” groups (
Effects of IGF-1 in rats kept in the cool recovery environment for the first 2 hours
Treatment with IGF-1 in the cool groups significantly reduced the overall percentage of neuronal loss compared with vehicle treatment (P < 0.05). There was no interaction between the time of the administration and effect of the treatment with IGF-1 or its vehicle. Post hoc testing for the time of administration suggested that treatment with IGF-1 (n = 28) at 2 hours after HI injury did not reduce the percentage of tissue damage (26.5% ± 36.3%) in the lateral cortex compared with its vehicle-treated group (n = 30, Fig. 5). The percentage of tissue damage in the vehicle-treated group was only 31.7% ± 37.5%. In contrast, IGF-1 (50 μg, n = 23) given 6 hours after HI injury significantly reduced the percentage of tissue damage in the lateral cortex (24.3% ± 35.3%) compared with the vehicle-treated group (46.7% ± 39.6%, n = 23, P < 0.05, Figs. 4C and D, and 5).
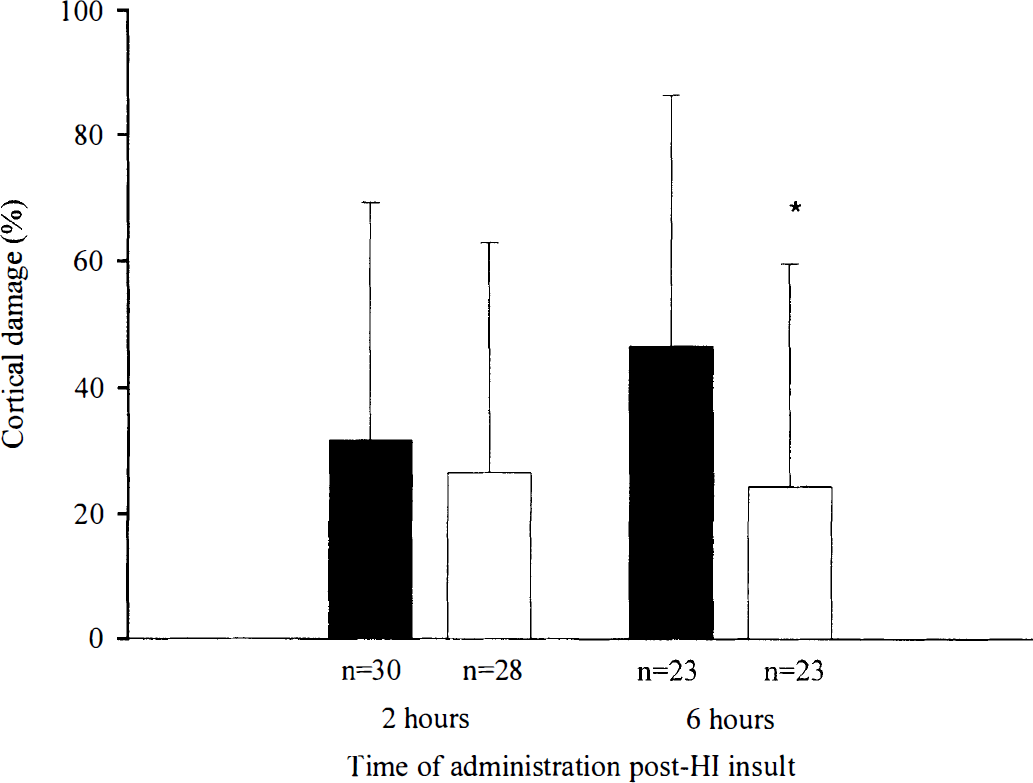
Effects of IGF-1 treatment on cortical injury in rats exposed to the “cool” recovery environment (23°C) for 2 hours after hypoxia—ischemia. In contrast to the “warm” group, IGF-1 did not reduce the percentage of cortical damage when administered centrally at 2 hours after hypoxia—ischemia. However, it significantly reduced cortical damage when given 6 hours after hypoxia—ischemia. *P < 0.05; data are presented as mean ± SD.
DISCUSSION
The current study demonstrates that the recovery environment in the first few hours after hypoxia—ischemia can critically modulate the window of opportunity for subsequent therapy. Cerebral temperature can fall sharply during HI and remain depressed for some time thereafter unless prevented by using a warm environment during this period (Miyazawa et al., 1993; Seif el Nasr et al., 1992). Consistent with previous reports (Sirimanne et al., 1996; Seif el Nasr et al., 1992), we found that cerebral temperature spontaneously fell during the first 2 hours of recovery after hypoxia in the normal (cool) holding environment. Under these conditions, cortical temperature was modestly reduced compared with either baseline recordings or the warm recovery group. There was a greater effect of the recovery environment on mean cerebral temperature (2.94°C) than on rectal temperature (0.86°C), emphasizing the need to measure cerebral temperature directly, as suggested by previous studies (Sirimanne et al., 1996).
After 2 hours' recovery in the warm environment, IGF-1 reduced cortical injury only when it was given 2 hours after hypoxia, consistent with our previous reports (Guan et al., 1996; Guan et al., 1993). There was no neuroprotective effect when the treatment was initiated later, at 6 hours after hypoxia. In contrast, after recovery in the cool (normal) environment, IGF-1 reduced cortical damage when given 6 hours after the HI injury. Thus, injury-induced cerebral cooling markedly prolonged the window of opportunity for effective treatment with IGF-1.
The timely selection and enrollment of patients for therapeutic trials in acute brain syndromes presents formidable logistic difficulties. Despite the proven efficacy of anticoagulation after ischemic stroke, for example, most patients would not able to be enrolled within its known narrow therapeutic window (Famularo et al., 1998). The current data suggest that posthypoxic mild hypothermia, particularly during early recovery, can prolong this window of opportunity for effective treatment while minimizing the known side effects associated with moderate hypothermia, including impaired immunocompetence and delayed coagulation, which may compromise recovery in very sick patients (Schubert, 1995).
Currently, the window of opportunity for any therapy can only be determined empirically. Some broad factors have been determined. Clearly, the initiation of neuronal apoptosis is accelerated by more severe insults. For example, DNA fragmentation in the hippocampus can be detected as early as 10 hours after a 60-minute HI injury in the rat, whereas DNA fragmentation in the hippocampus is detectable only 3 to 5 days after a 15-minute HI injury (Beilharz et al., 1995). However, DNA fragmentation and classic ischemic cell change represent only the terminal events of this cascade; thus, their appearance is not a good guide as to when cell death still may be reversible.
In vitro studies have distinguished a latent and an active or “execution” phase of programed cell death, each with a different cytoplasmic biochemical profile (Samejima et al., 1998). Whereas the active phase showed the presence of several downstream factors that could induce DNA fragmentation and chromatin condensation within substrate nuclei, the preceding latent phase was characterized by activated caspases without the downstream factors. These data suggest that activation of the intranuclear factors is the critical event that occurs at the transition from the latent to active phases of programed neuronal death.
The mechanism of neuronal rescue with IGF-1 is believed to be related to suppression of events in this latent phase of apoptosis (D'Mello et al., 1993). IGF-1 suppresses apoptosis through mechanisms involving the phosphatidylinositol 3'-kinase and mitogen-activated protein kinase pathways (Parrizas et al., 1997). These pathways lead to inhibition of the caspase cascade directly or indirectly (Tamatani et al., 1998). The inhibition of cell death is broadly based, with evidence that IGF-1 can block otherwise neurotrophin-insensitive apoptosis (Fernandezsanchez et al., 1996).
The mechanisms of change in postinsult temperature on cell death remain unclear. Hypothermia can suppress release of excitotoxins, nitric oxide, and free radicals during reperfusion (Lei et al., 1997; Thoresen et al., 1997). Also, there is evidence that mild postinsult hypothermia has a specific effect to inhibit apoptotic cell death and cellular DNA fragmentation in several species (Xu et al., 1998; Edwards et al., 1995). Short-term cerebral cooling during brain recovery may not be neuroprotective (Sirimanne et al., 1996; Gunn, Gunn, 1998); however, it is reported to delay the onset of secondary neuronal death (Colbourne et al., 1998; Gunn et al., 1998; Trescher et al., 1997). The current data, which show that a small (2.9°C) change in brain temperature greatly alters the therapeutic window, are consistent with this hypothesis and suggest that the hypothermia is suppressing the evolution of the latent phase of programed neuronal death.
Interestingly, in the cool recovery environment group, IGF-1 did not significantly improve neuronal outcome when given 2 hours after the injury. This must be interpreted with caution, however, since the overall treatment effect with IGF-1 showed no interaction with time of administration. If confirmed in further studies, this would suggest that concurrent hypothermia might attenuate in some way the effect of IGF-1, or that the deleterious processes that IGF-1 can inhibit had not yet been activated. We have previously shown that, for example, IGF-1 was not effective when administered 1 hour before hypoxemia—ischemia, raising the possibility that administration may be effective only once certain processes have commenced (Gluckman et al., 1998; Gluckman et al., 1992).
CONCLUSION
Spontaneous mild, brief cerebral cooling after hypoxemia—ischemia was not neuroprotective by itself; however, it markedly prolonged the window of opportunity for effective treatment with IGF-1. Recovery for 2 hours in the cool environment instead of the warm environment enabled neuroprotection with IGF-1 as late as 6 hours after the initial brain injury, speculatively, by delaying the evolution of the latent phase of programed neuronal death. These data indicate that the timing of effective neuronal rescue therapies may be critically modulated by postinsult cerebral temperature.