
Abstract
Both thrombin and plasmin induce contraction of brain endothelial cells, which may increase capillary permeability thereby leading to disruption of the blood-brain barrier. Identification of thrombin receptors, as well as the influence of plasmin on their activation, in capillary endothelial cells and astrocytes are therefore essential for understanding injury-related actions of thrombin in the brain. Using the reverse transcriptase-polymerase chain reaction method, the present study shows that primary cultures of rat brain capillary endothelial (RBCE) cells and astrocytes derived from rat brain express two different thrombin receptors. The first is proteolytically activated receptor (PAR)-1, the receptor responsible for the vast majority of the thrombin's cellular activation functions; the second is PAR-3, a receptor described to be essential for normal responsiveness to thrombin in mouse platelets. In addition to these thrombin receptors, the mRNA (messenger RNA) for PAR-2, a possible trypsin receptor, was also identified. Functional significance of thrombin receptors was indicated by changes in [Ca2+]i in response to thrombin, as measured by FURA-2 fluorescence in RBCE cells. Thrombin as low as 4 nmol/L induced an abrupt increase in [Ca2+]i whereas, upon addition of active site-blocked thrombin or plasmin, [Ca2+]i remained unchanged. The [Ca2+]i signal attributable to thrombin was smaller in a low Ca2+-containing medium, indicating that an influx of Ca2+ from the extracellular medium makes a contribution to the overall [Ca2+]i rise. The amplitude of the transient [Ca2+]i signal was dependent on the concentration of thrombin, and repeated application of the enzyme caused an essentially complete and long-term desensitization of the receptor. The PAR-1 agonist peptide SFLLRN also elicited a transient increase in [Ca2+]i. After activation by SFLLRN, cells showed a diminished response to thrombin, but the response was not absent, indicating that PAR-3 might contribute to the generation of the [Ca2+]i signal. Pretreatment of RBCE cells with 100 nmol/L plasmin completely prevented [Ca2+]i rise attributable to thrombin. These data show that RBCE cells and astrocytes express at least two receptors for thrombin, PAR-1 and PAR-3, and probably both receptors are involved in thrombin-induced [Ca2+]i signals. Plasmin itself does not elevate [Ca2+]i but prevents the activation of receptors by thrombin.
Thrombosis is of paramount importance in the pathophysiology of ischemic stroke. Thrombogenic and fibrinolytic enzymes accumulating in the occluded vascular segments may play a role in the opening of the blood-brain barrier, and the subsequent exposure of cells in the brain to high amounts of thrombin and plasmin is likely to have deleterious effects. Thrombin actions are largely mediated by PAR-1, the first member of the proteolytically activated G-protein-coupled receptor family to be discovered (Vu et al., 1991; Rasmussen et al., 1991). The N-terminus of this receptor is cleaved by thrombin between residues Arg41 and Ser42, exposing a new N-terminus whose first five residues serve as a tethered ligand initiating the receptor activation. Accordingly, synthetic peptides comprising the amino terminus of the cleaved receptor, such as SFLLRN, can mimic the effects of thrombin. Thrombin receptors homologous to those on platelets have also been identified on fibroblasts (Rasmussen et al., 1991; Coughlin et al., 1992), endothelial cells (Vu et al., 1991; Bartha et al., 1993; Molino et al., 1995), smooth muscle cells (Zhong et al., 1992), and macrophages (Coughlin et al., 1992).
PAR-1 mRNA (messenger RNA) is expressed in high levels in the nervous system (Soifer et al., 1994; Suidan et al., 1996), in neuronal, glial, and ependymal cells (Weinstein et al., 1995). Thrombin can affect a number of functions in the brain; it causes neurit retraction (Gurwitz and Cunningham, 1988), stimulates proliferation in astrocytes (Perraud et al., 1987), and increases the expression of nerve growth factor in glial cells (Neveu et al., 1993). In rat C6 glioma cells thrombin provokes [Ca2+] signals (Tas and Koschel, 1990). A strong decrease in thrombin receptor expression occurs in most parts of the brain between the second and third postnatal week (Niclou et al., 1994), suggesting that the receptor may be important for development (Risau and Wolburg, 1990). Additional circumstantial evidence supporting the importance of thrombin receptors in the brain comes from studies demonstrating the expression of prothrombin mRNA (Dihanich et al., 1991), its regional codistribution with PAR-1 (Soifer et al., 1994), and the presence of the thrombin inhibitor protease nexin around cerebral blood vessels where the inhibitor could protect neural tissue from extravasated thrombin (Choi et al., 1990).
Endothelial cells of brain capillaries are the site of the blood—brain barrier, which guarantees brain homeostasis by preventing free diffusion of molecules into the brain (Pardridge, 1991). Intimately associated with endothelial cells, astrocytes are believed to participate in the blood-brain barrier function; they contribute to tight junction formation (Janzer and Raff, 1987). In addition, astrocytes provide a trophic support for neuronal growth and differentiation (Vernadakis, 1988). In astrocytes effects of thrombin have been shown to be mediated by PAR-1 (Pike et al., 1996), but data on thrombin receptor mRNA as well as on thrombin-induced alterations in brain capillary endothelial cells are contradictory; in fact the presence of functional thrombin receptors in these cells has not yet been demonstrated. Although it has been reported that PAR-1 mRNA is widely expressed in embryonic mouse brain capillary endothelial cells (Soifer et al., 1994), in situ hybridization studies have failed to show the presence of PAR-1 mRNA in the cerebrovascular endothelium derived from adult rat brain (Weinstein et al., 1995). Apparently inconsistent with the latter finding, thrombin causes shape changes (Nagy et al., 1995) and stimulates urokinase production and DNA synthesis (Shatos et al., 1995) in cultured human brain capillary endothelial cells. It is possible that thrombin receptors different from PAR-1 are responsible for these cellular effects, since recent studies have provided evidence for the existence of a more extended gene family of PAR-s displaying similar mechanism of activation. PAR-2 is known to be activated by trypsin and tryptase but not by thrombin (Nystedt et al., 1995). In addition to responding to peptide agonists based upon its own tethered ligand domain, PAR-2 can also be activated by the PAR-1 agonist peptide SFLLRN (Molino et al., 1997). In mouse and rat platelets another thrombin receptor, PAR-3, is necessary for normal thrombin responses (Ishihara et al., 1997), whereas PAR-1 plays no apparent role (Connolly et al., 1994). Furthermore, besides PAR-3, a PAR-4-mediated mechanism for thrombin signaling has also been demonstrated in these cells (Kahn et al., 1998).
Any protease can be an agonist on protease-activated receptors which cleave the N-terminus at the site required for the exposure of the tethered ligand domain. Plasmin can bind with high affinity to endothelial cells (Bauer et al., 1984; Hall et al., 1991), eliciting functional responses through a specific plasmin receptor, but by cleaving at Arg41-Ser42 it also has the potential to activate PAR-1 (Parry et al., 1996). Plasmin is known to affect platelet activation; at high concentrations it increases, but at lower concentrations it inhibits thrombin-induced platelet activation (Kimura et al., 1996; Kinlough-Rathbone et al., 1997). Plasmin also modulates the thrombin-evoked calcium response in C6 glioma cells (Turner et al., 1994). Recently we have shown that not only thrombin but also plasmin can induce the contraction of human brain capillary endothelial cells in culture (Nagy et al., 1995). Because thrombin receptors have not yet been identified in brain capillary endothelial cells, the present study aims to show the expression of the receptors in primary cultures of these cells derived from rat brain and to compare them with the receptors present on astrocytes. In order to demonstrate functional responses of the cells, we measured the changes in [Ca2+]i after stimulation with thrombin or the PAR-1 agonist peptide SFLLRN and examined the modification of the thrombin-induced changes in [Ca2+]i by plasmin.
METHODS
Preparation of rat brain capillary endothelial cells
Endothelial cells were obtained from brain capillary fragments using a method described earlier (Dömötör et al., 1998). Shortly, the gray matter of the brain from 3- to 5-month-old Lewis rats was chopped into 2- to 3-mm pieces, homogenized, and centrifuged to separate capillary fragments. The capillary pellet was digested for 3 hours in a 0.1% collagenase/dispase (Boehringer-Mannheim, Germany) solution, and the digested suspension was centrifuged on a Percoll (Pharmacia, Uppsala, Sweden) density gradient. The isolated capillary fragments were plated on extracellular matrix-coated glass coverslips which were prepared as described previously (Dömötör et al., 1998).
Brain endothelial cell cultures plated on extracellular matrix were kept in DMEM containing 17% plasma-derived bovine serum (Sigma), supplemented with 2 mmol/L glutamin, 80 μg/ mL heparin, 150 μg/mL endothelial cell growth supplement (Sigma, St. Louis, MO, U.S.A.), antibiotics, and trace factors (vitamin C, selenium, insulin, transferrin, and glutathione).
To obtain pure endothelial cultures, cells were exposed to anti-Thy 1.1-complement killing procedure (Dömötör et al., 1998). On the second day after seeding, cells were incubated with monoclonal anti-Thy 1.1 antibody (Sigma, 1:500 dilution in serum-free medium) for 40 minutes at 37°C. After washing, rabbit complement (Sigma, dilution 5:3 in serum-free medium) was added for a maximum of 120 minutes at 37°C in order to lyse pericytes and astrocytes. After washing, the cells were normally fed with serum-containing medium. Cell cultures became confluent after 7 to 10 days. Only primary cultures were used for the experiments.
Preparation of primary astrocyte culture
Astrocyte culture was prepared from forebrain of 1-day-old rats as described by Madarász et al. (1991).
Immunocytochemistry
Von-Willebrand antigen was detected on cells permeabilized with 0.4% paraformaldehyde and Triton X-100. Cells were incubated with rabbit anti-human Von-Willebrand factor antibody (factor VIII-related antigen, Dako), and this was followed by incubation with biotinilated anti-rabbit IgG and the streptavidin-peroxidase complex (Dako, Glostrup, Dania).
Measurement of [Ca2+]i in single cells
[Ca2+]i was measured as detailed elsewhere (Dömötör et al., 1999). Endothelial cells plated on extracellular matrix-coated coverslips were loaded with Fura-2 acetoxymethyl ester (Sigma) for 1 hour at 37°C in culture medium, followed by the transfer of the coverslips into a preformed perfusion chamber fitted to the stage of an inverted microscope. The perfusion chamber was prewarmed to 37°C, and the cells were superfused with a Hepes buffer of the following composition (in mmol/L): 141.6 NaCl, 5.4 KCl, 1.6 MgCl2, 2 CaCl2, 11 glucose, 20 Hepes, pH 7.4, at a rate of 2 mL/min for at least 10 minutes before the experiments. Agonists were introduced using a very fine perfusion system; the diameter of the input tip was constructed to be approximately 60 to 100 μm and was adjusted before the experiments to the close vicinity of the particular cell with a micromanipulator under microscope control.
Excitation wavelengths were 343 and 376 nm (bandwidth, 10 nm). Fluorescence ratio of F343 over F376 was monitored using a PTI PatioMaster System equipped with Felix for Windows Softwares, and converted to free [Ca2+]i by the equation of Grynkiewicz et al. (1985) using an in vitro calibration method and a correction for the viscosity (Poenie, 1994). Low Ca2+ solutions (~50 nmol/L) were made with 0.5 mmol/L CaCl2 and 1.2 mmol/L EGTA.
RNA extraction and polymerase chain reaction analysis
At confluence, the cell monolayers were washed with phosphate-buffered saline, and total RNA was isolated by the RNA NOW procedure (Biogentex, Montigny, France). The RNA pellet was extracted by ethanol, dried, dissolved in 10 μL of water, and RNA concentration was determined by absorbance at 260 nm. Total RNA (100 ng) was reverse-transcribed (50°C for 30 minutes) and amplified using the Titan RT-PCR (Boehringer) system. Polymerase chain reaction conditions were 30 cycles, each consisting of 1 minute of denaturation at 94°C, 1 minute of hybridization at 57°C, and 2 minutes of elongation at 72°C. The amplified products were separated by 1.2% agarose gel electrophoresis, and the bands were visualized using ethidium bromid. The sequences for sense and antisense primers for the rat PAR-1 were 5, ACTATTTCTCCGCCTTCTCCGCCAT 3, corresponding to nucleotides 900-924, and 5, TCACGCAGACGCAGAGGAGGAGGTAAGC 3, corresponding to nucleotides 1148–1172 (Zhong et al., 1992). The sequences for the rat PAR-2 primers were 5, AACCTGGCCTTGGCAGACCTCCTCTCT 3, corresponding to nucleotides 346-372, and 5, ATGCGTTTCACGGTGCGGACGCTT 3, corresponding to nucleotides 1086–1109 (Saifeddine et al., 1996). The sequences for PAR-3 primers: 5, CTTTTCTGTGTCACGCTGCCG 3, and 5, ACAGGTGAGGATGAGAATAGC 3, were from the rat platelet cDNA.
Purification of thrombin and plasminogen
Thrombin was purified from crude bovine thrombin (Sigma, 50 NIHU/mg protein) with ion-exchange chromatography (Lundblad et al., 1976); the specific activity of the thrombin after purification was 1,150 NIHU/mg protein.
Active-site blocked thrombin was prepared by incubating thrombin with 4 mmol/L Pefablock (Boehringer) for 30 minutes. Thrombin concentration was determined by active site titration using p-nitrofenilguanidinobenzoate.
Plasminogen was prepared from human plasma by affinity chromatography on lysine-sepharose (Deutsch and Metz, 1970). Plasmin was prepared from zymogen by activation with streptokinase; the concentration of the active enzyme was determined using Spectrozyme-PL (American Diagnostica, New York, U.S.A.).
Statistics Comparison of means was made by using the Student's t test for paired samples.
RESULTS
Identification of brain capillary endothelial cells
Endothelial cells were spindle-shaped and grew in close contact with each other within 5 to 10 days (Fig. 1A). The endothelial character of these cells was confirmed by their strong positive granular, perinuclear immunoperoxidase staining for von-Willebrand antigen (Fig. 1B).
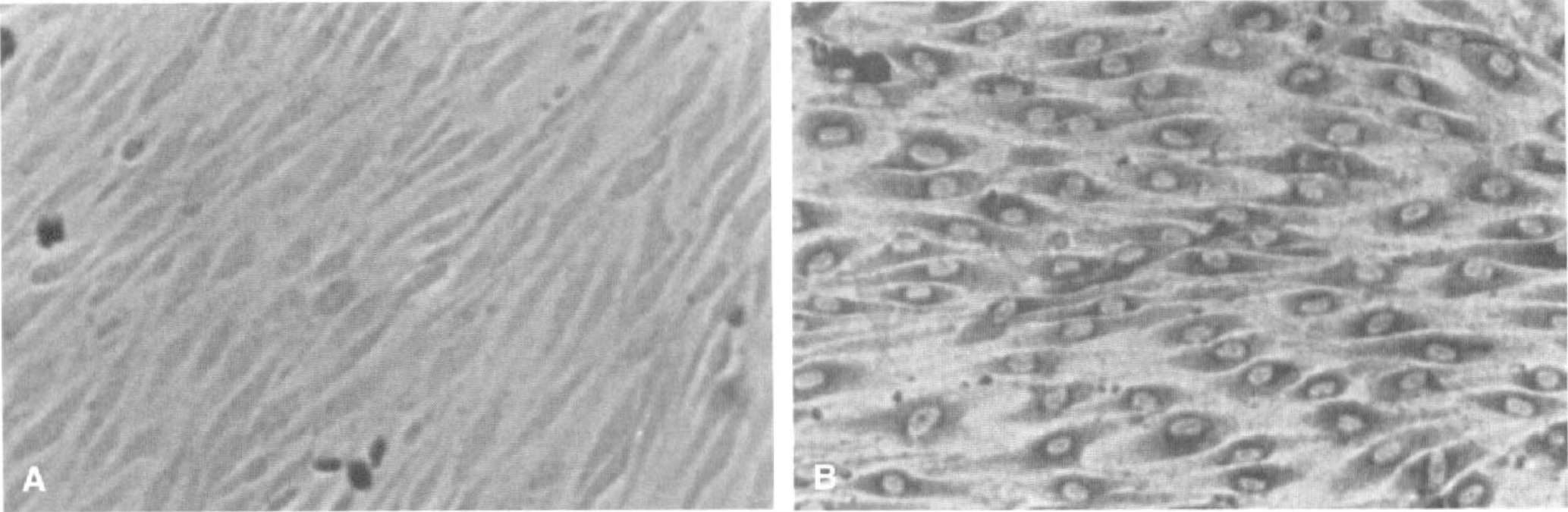
Characteristics of cultured rat brain capillary endothelial (RBCE) cells by phase-contrast microscopy.
Thrombin receptor expression in rat brain
Reverse transcriptase polymerase chain reaction using PAR-1, PAR-2, and PAR-3 primers was performed to detect the presence of thrombin receptors on primary cultures of capillary endothelial cells and astrocytes prepared from rat brain, as described in Methods above. Rat aortic smooth muscle cells, known to express PAR-1 and PAR-2, were used as a positive control. Primers were designed to rat PAR-1 (Zhong et al., 1992), PAR-2 (Saifeddine et al., 1996), and PAR-3 sequences, respectively. Both PAR-1 and PAR-3 thombin receptor mRNA and also PAR-2 mRNA were detected in the cDNA of rat brain capillary endothelial (RBCE) cells, as well as in that of astrocytes (Fig. 2).
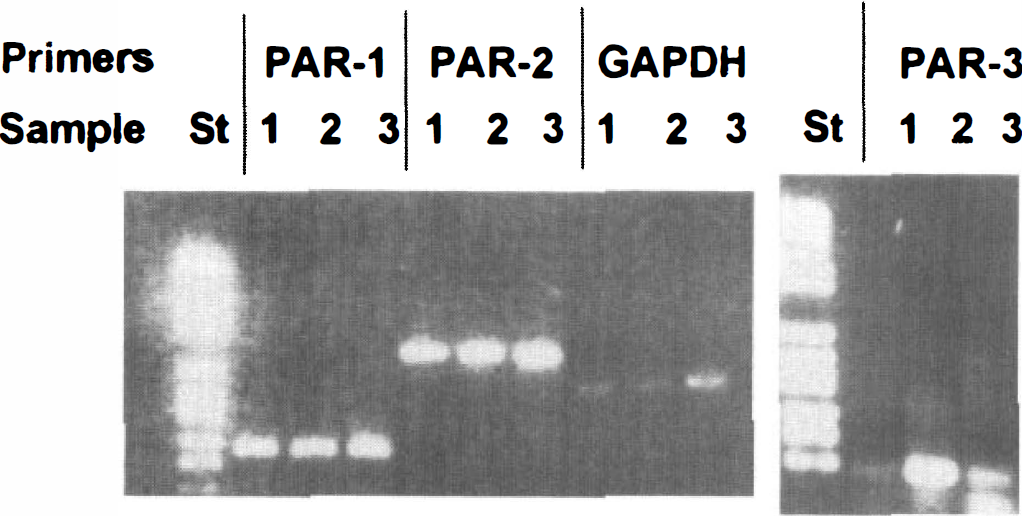
Expression of proteolytically activated receptor (PAR)-1, PAR-2, and PAR-3 messenger RNA (mRNA) in rat brain capillary endothelial (RBCE) cells. Total cellular RNA was isolated from confluent monolayers of RBCE cells; 100 ng was reverse-transcribed and subsequently amplified using proteolytically activated receptor (PAR)-1, PAR-2, PAR-3, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers. The molecular size was determined using DNA molecular weight marker VI, and the marker (St) is shown beside sample 1 amplified using PAR-1 and PAR-3 primers. Polymerase chain reaction (PCR) products of PAR-1, PAR-2, PAR-3, and GAPDH mRNA from RBCE cells (sample 1), from astrocytes of rat forebrain (sample 2) and from rat smooth muscle cells (sample 3), respectively, are shown.
Effect of thrombin on [Ca2+]i of rat brain capillary endothelial cells
Figure 3 (inset) demonstrates that treatment of RBCE cells with 40 nmol/L thrombin (2 NIHU/mL) resulted in a sharp, transient increase of [Ca2+]i, reaching a maximum within 12 seconds which then gradually declined (within 180 seconds) to baseline. When the same experiment was performed in a medium containing Ca2+ in low concentration, the extent of the thrombin-induced increase in [Ca2+]i was reduced, indicating that the rapid changes in [Ca2+]i were, at least partly, dependent on the presence of extracellular Ca2+ (data not shown). The effect of thrombin on [Ca2+]i was concentration-dependent, as demonstrated in Fig. 3.
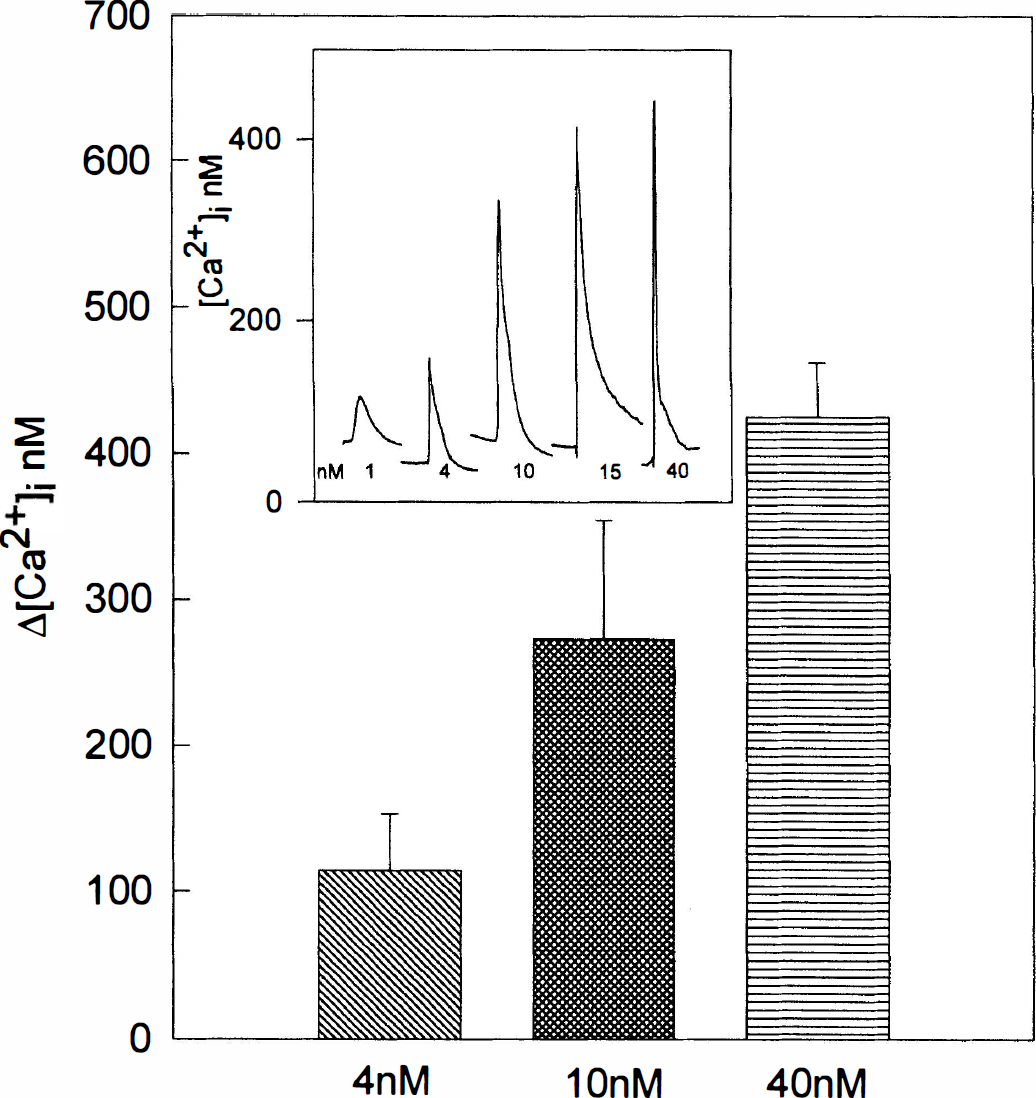
Dose dependency of the action of thrombin on [Ca2+]i. Rat brain capillary endothelial (RBCE) cells were stimulated with thrombin for 60 seconds at concentrations indicated. Δ[Ca2+]i is expressed as the difference between peak [Ca2+]i and baseline [Ca2+]i measured in three experiments using cells from three different preparations (mean ± SD). Resting [Ca2+]i was 52 ± 6 nmol/L (n = 25). The inset illustrates individual [Ca2+]i signals initiated by thrombin at concentrations indicated.
The requirement of a cleavage event for the [Ca2+]i rise in response to thrombin in RBCE cells was confirmed by the use of an active-site blocked thrombin which was without an effect (data not shown). Agonist-dependent desensitization of the thrombin response was examined by exposing RBCE cells to two consecutive thrombin stimuli. Repeated addition of thrombin (40 nmol/L) 300 seconds after the first stimulus induced a very small [Ca2+]i signal (Fig. 4), demonstrating rapid homologous desensitization. Thrombin responsiveness did not increase within the next 2 hours (data not shown), and because cleaved receptors are unable to respond to thrombin this result indicated that new receptors were not expressed in the cells within this period.
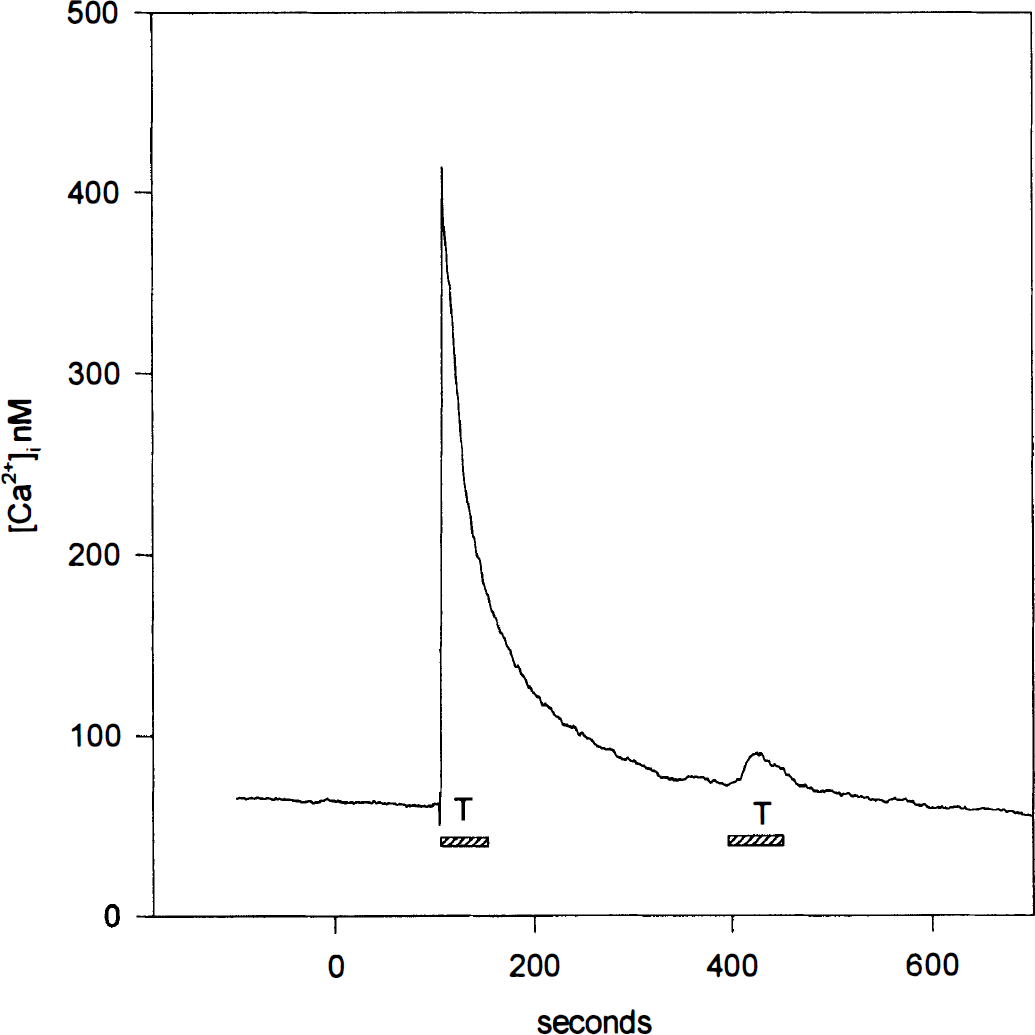
Desensitization of the thrombin-induced [Ca2+]i response by thrombin. Rat brain capillary endothelial (RBCE) cells were stimulated with 40 nmol/L thrombin for 60 seconds as indicated (shaded rectangle). After 300 seconds they were again exposed to the same concentration of thrombin. Traces are representative of two experiments performed on two different preparations.
Effects of SFLLRN on [Ca2+]i
Like thrombin, SFLLRN also elicited a transient increase in [Ca2+]i. However, the overall response to SFLLRN was slower and smaller in magnitude as compared to the increase in [Ca2+]i induced by thrombin (Fig. 5). Peak [Ca2+]i rise attributable to 100 μmol/L SFLLRN was only half of the increase induced by 40 nmol/L thrombin. In contrast to the essentially complete desensitization by thrombin (Fig. 4), SFLLRN-stimulated cells were still able to respond to a thrombin-stimulus, albeit with an attenuated [Ca2+]i signal (Fig. 5). The magnitude of the [Ca2+]i increase attributable to thrombin appeared to inversely correlate with the concentration of the first stimulus: cells activated with 100 μmol/L SFLLRN responded to thrombin with a smaller [Ca2+]i signal compared to those stimulated with 50 μmol/L agonist peptide (=40% and 60% of the response induced by thrombin alone, respectively).
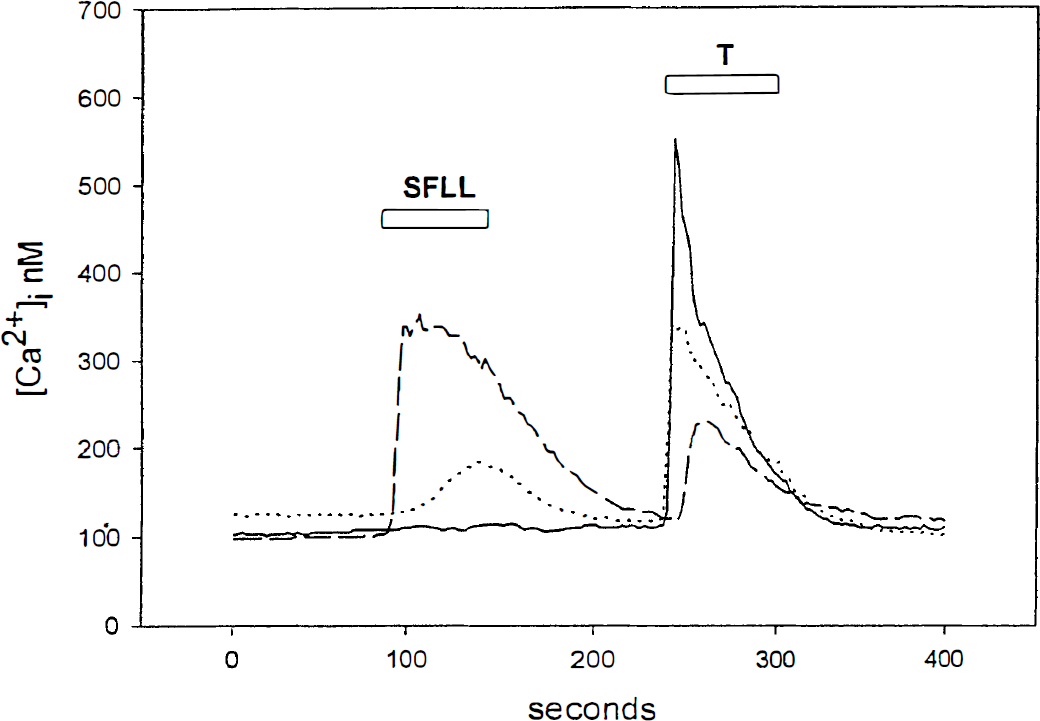
Effect of SFLLRN on [Ca2+]i. [Ca2+]i in endothelial cells incubated for 60 seconds in the absence (−) or in the presence of 50 μmol/L (…) or 100 μmol/L (−) SFLLRN, then stimulated with 40 nmol/L thrombin (T) as indicated. The results are representative of five experiments performed on three different preparations.
Modification of thrombin responses by plasmin
We have previously shown that plasmin, as well as other fibrinolytic enzymes (urokinase and elastase), provoked a contraction of human brain capillary endothelial cells (Nagy et al., 1995). In the present study we investigated the effect of plasmin on [Ca2+]i in RBCE cells. When exposed to plasmin (10 to 400 nmol/L) for up to 5 minutes no increase in [Ca2+]i was observed (not shown). However, pretreatment of cells with plasmin (100 nmol/ L) almost completely abolished [Ca2+]i rise in response to thrombin (Fig. 6A), suggesting that plasmin prevented receptor activation by thrombin. Although plasmin completely abolished the activation of RBCE cells by thrombin, it failed to influence the [Ca2+]i signal induced by ATP, a known purinergic agonist (Fig. 6B). This indicates that the effect of plasmin on the thrombin-induced [Ca2+]i rise is specific and involves interaction with the thrombin receptor.
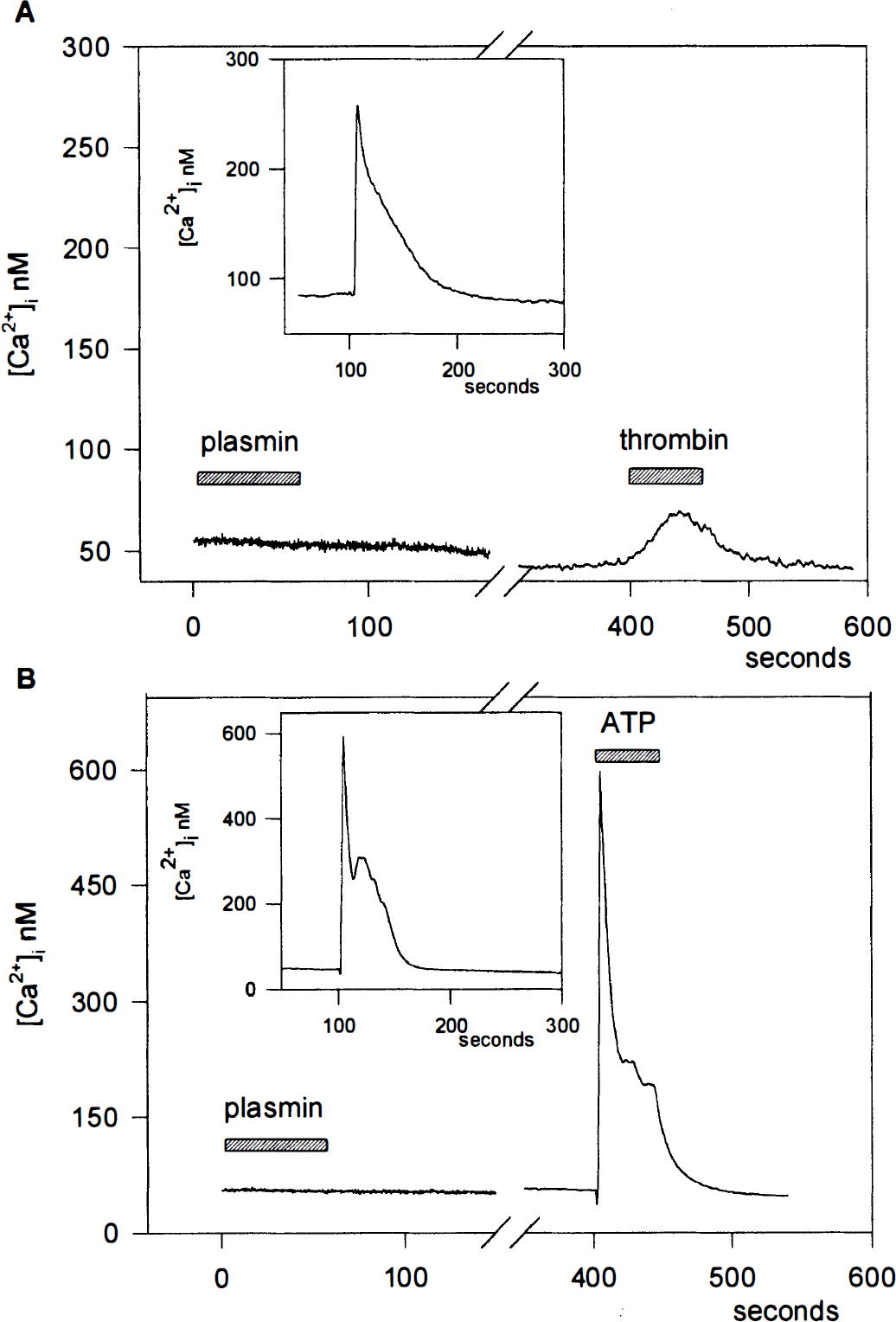
Effect of plasmin pretreatment on the thrombin- and the ATP-induced [Ca2+]i.
DISCUSSION
In the present study we demonstrate for the first time the presence of thrombin receptor mRNA and functional thrombin receptors in capillary endothelial cells of the brain (Fig. 2). One of the receptors identified is PAR-1, which is commonly expressed in platelets (Vu et al., 1991; Hung et al., 1992) and large-vessel endothelial cells (Vu et al., 1991; Hein et al., 1994) but has not yet been found in brain capillary endothelial cells. Stimulation of PAR-1 by thrombin resulted in an increase of [Ca2+]i in RBCE cells (Fig. 3). In the presence of a low concentration of extracellular Ca2+, the thrombin-induced signal was reduced but not abolished, which suggests that both influx of Ca2+ from the medium and release of Ca2+ from intracellular pools are involved. In addition to thrombin, the PAR-1 agonist peptide SFLLRN also elicited an increase in [Ca2+]i. Results in Fig. 5 show that SFLLRN diminished but did not completely eliminate the subsequent [Ca2+]i response to thrombin and that the magnitude of the [Ca2+]i signal attributable to thrombin was inversely related to the concentration of SFLLRN applied previously. Recent data have shown that, in addition to acting on PAR-1, SFLLRN can activate at least one other receptor: PAR-2, also present on the endothelial cell surface but not activated by thrombin (Nystedt et al., 1995). Consistent with this, in human umbilical vein endothelial cells, [Ca2+]i response to SFLLRN is larger than that induced by thrombin (Molino et al., 1997). Although we show that RBCE cells as human umbilical vein endothelial cells express PAR-2 (Fig. 2), the persistently higher [Ca2+]i signal induced by thrombin as compared to that elicited by SFLLRN suggests the coexistence of another thrombin receptor in RBCE cells which could not be activated by the PAR-1 agonist peptide. Indeed, this paper demonstrates the presence of PAR-3 mRNA in RBCE cells, which could contribute to the [Ca2+]i rise in response to thrombin but not to SFLLRN. As with endothelial cells, astrocytes derived from rat forebrain also expressed PAR-3, in addition to PAR-1 and PAR-2 (Fig. 2). Thrombin has been reported to reverse the β-amyloid-induced stellate morphology in astrocytes, and this effect has been mimicked by the PAR-1 agonist peptide (Pike et al., 1996). In contrast, thrombin potentiates rather than attenuates the β-amyloid-induced b-FGF secretion (Pike et al., 1996), suggesting that it differentially modulates the effects of β-amyloid in astrocytes and that the effects might be mediated through different thrombin receptors.
According to earlier findings, PAR-1 becomes quickly desensitized after activation by thrombin (Brass, 1992; Ishii et al., 1994). This is thought to be attributable to two events: phosphorylation of sites in the cytoplasmic domains of the receptor and cleavage of the receptor N-terminus (Ishii et al., 1994). However, numerous studies addressing PAR-1 regulation by thrombin indicate that it is highly variable and cell type-specific (Weinstein et al., 1998). In platelets and megakaryoblastic cell lines, receptor cleavage is followed by rapid internalization with a full recovery of the thrombin responsiveness occurring over a long time period (Hein et al., 1994). In contrast, in endothelial cells and in fibroblasts, an intracellular pool of intact receptors rapidly replaces the cleaved receptors and allows cells to respond to the next stimulus (Hein et al., 1994). In neuronal cells, PAR-1 regulation by thrombin resembles that observed in megakaryocytes (Weinstein et al., 1998). Here we report that RBCE cells also became refractory to a second stimulation by thrombin (Fig. 4) and no recovery of the thrombin responsiveness was observed for at least 2 hours. These observations indicate that RBCE cells do not restore intact thrombin receptors at a rate comparable to that in human umbilical vein endothelial cells; the pattern of slow receptor recovery was similar to that reported for platelets, megakaryoblasts, and neurons. Conversely, when RBCE cells were initially activated by a high concentration of SFLLRN, they showed a diminished but still measurable response to thrombin (Fig. 5), even when the peptide was not washed out from the incubation medium. This observation is again markedly different from that made in human umbilical vein endothelial cells, where PAR-1 was desensitized after receptor activation by either thrombin or SFLLRN. Because PAR-3 is not activated by SFLLRN, this result could be explained by the coexpression of PAR-1 and PAR-3 receptors on RBCE cells. The desensitization of PAR-3 by thrombin has not yet been studied, but it may be different from that of PAR-1.
The experiments on a possible effect of plasmin on [Ca2+]i in RBCE cells were motivated by our previous observation that nanomolar concentrations of plasmin caused contractions in cultured human brain capillary endothelial cells (Nagy et al., 1995), and by the fact that thrombin receptor was cleaved by plasmin at Arg41-Ser42 (Parry et al., 1996). We found that plasmin itself did not cause any increase in [Ca2+]i, indicating that plasmin-induced contraction in endothelial cells might be attributable to a Ca2+-independent mechanism. In addition, plasmin was able to abolish the thrombin-induced increase in [Ca2+]i (Fig. 6). This is in agreement with results obtained in platelets (Kimura et al., 1996) and C6 glioma cells (Turner et al., 1994), where low doses of plasmin inhibited thrombin-induced responses. Probably plasmin, besides cleaving PAR-1 at Arg41-Ser42, also cleaves the receptor at a site closer to the C terminus resulting in the loss of the tethered ligand, as has been shown for cathepsin G (Molino et al., 1995). As yet there are no data on the cleavage of PAR-3 by plasmin. Our data showing that plasmin completely abolishes the thrombin-induced increase in [Ca2+]i in RBCE cells, where both PAR-1 and PAR-3 are present, would suggest that plasmin also inactivates PAR-3, but further experiments are needed to prove this.
Reverse transcriptase-polymerase chain reaction analysis in our experiments also showed the presence of PAR-2 in RBCE cells and in astrocytes. PAR-2 is known to be activated by trypsin, or mast cell tryptase; however, the native physiologic activator and the cellular function of PAR-2 remain unknown. A recent study has provided evidence that PAR-2 is also present in the brain, and as is the case with PAR-1, it may play a role in neurodegeneration (Smith-Swintosky et al., 1997). Our results, showing the presence of PAR-2 mRNA in primary cultures of astrocytes and capillary endothelial cells from rat brain, support these data and also suggest a role for PAR-2 in the brain.
In conclusion, PAR-1 mRNA is expressed in RBCE cells, and stimulation of PAR-1 receptors by thrombin and by SFLLRN mediates an increase in [Ca2+]i. However, another thrombin receptor, PAR-3, is also expressed and might contribute to the thrombin-induced increase in [Ca2+]i. The susceptibility of the receptor(s) to proteolysis by plasmin may play a role in the response of the endothelial cell to thrombin, when fibrinolysis occurs parallel with blood coagulation. Besides playing a principal role in hemostasis, thrombin may be an important regulator in neuronal development and may contribute to brain injury in certain pathologic situations. The identification of PAR-1 and PAR-3 in endothelial cells and astrocytes may help to elucidate the role and importance of distinct thrombin receptors in the brain.
Footnotes
Acknowledgments
The authors thank Gy. Oravecz for expert assistance and Mark L. Kahn (University of California, San Francisco) for kindly providing the sequence of the rat PAR-3.