
Abstract
The authors investigated the role of the prostaglandin-synthesizing enzyme cyclooxygenase-2 (COX-2) in the mechanisms of focal cerebral ischemia and its interaction with inducible nitric oxide synthase (iNOS). Focal cerebral ischemia was produced by permanent occlusion of the middle cerebral artery (MCA) in mice. Infarct volume was measured 96 hours later by computer-assisted planimetry in thionin-stained brain sections. The highly selective COX-2 inhibitor NS398 (20 mg/kg; intraperitoneally), administered twice a day starting 6 hours after MCA occlusion, reduced total infarct volume in C57BL/6 (–23%) and 129/SVeV mice (–21%), and ameliorated the motor deficits produced by MCA occlusion (P < .05). However, NS398 did not influence infarct volume in mice with deletion of the iNOS gene (P > .05). In contrast, the neuronal NOS inhibitor 7-NI (50 mg/kg; intraperitoneally), administered once 5 minutes after MCA occlusion, reduced neocortical infarct volume by 20% in iNOS −/− mice (P < .05). NS398 did not affect arterial pressure, resting CBF or the CBF reactivity to hypercapnia in anesthetized iNOS null mice (P > .05). The data suggest that COX-2 reaction products, in mouse as in rat, contribute to ischemic brain injury. However, the failure of NS398 to reduce infarct volume in iNOS null mice suggests that iNOS-derived NO is required for the deleterious effects of COX-2 to occur. Thus, COX-2 reaction products may be another mechanism by which iNOS-derived NO contributes to ischemic brain injury.
Keywords
Focal cerebral ischemia, in rodents as in humans, is associated with induction of the immunologic isoform of inducible nitric oxide synthase (iNOS) (Iadecola, 1997; Samdeni et al., 1997). iNOS is normally not present in most cells, but its expression is transcriptionally induced by inflammatory stimuli (MacMicking et al., 1997). At variance with neuronal and endothelial NOS, iNOS produces micromolar amounts of NO continuously and mediates cytotoxicity (Dawson et al., 1994; Hewett et al., 1994; Hewett et al., 1996; Vodovotz et al., 1994). After middle cerebral artery (MCA) occlusion in rodents, iNOS is expressed in infiltrating neutrophils and in vascular cells throughout the ischemic territory (Iadecola et al., 1996; Iadecola et al., 1995b). Maximal iNOS mRNA expression occurs 48 hours after permanent MCA occlusion (Iadecola et al., 1995b). Administration of the relatively selective iNOS inhibitor aminoguanidine, starting 12 to 24 hours after ischemia, attenuates iNOS activity in the ischemic brain, reduces cerebral ischemic damage, and ameliorates the motor deficits produced by cerebral ischemia (Iadecola et al., 1995a; Nagayama et al., 1998). Furthermore, focal cerebral ischemic damage is reduced in mice with deletion of the iNOS gene (Iadecola et al., 1997). These findings, collectively, suggest that NO produced by iNOS contributes to ischemic brain injury.
Another inflammation-related gene that is expressed after cerebral ischemia is the prostaglandin-synthesizing enzyme cyclooxygenase-2 (COX-2) (Collaco-Moraes et al., 1996; Miettinen et al., 1997; Nogawa et al., 1997; Planas et al., 1995). After MCA occlusion, COX-2 expression is upregulated with a time course similar to that of iNOS (Nogawa et al., 1997). However, at variance with iNOS, COX-2 is expressed not only in vascular cells, but also in injured neurons at the infarct's border (Miettinen et al., 1997; Nogawa et al., 1997). Administration of the COX-2 inhibitor NS398 attenuates the increase in prostaglandins induced by cerebral ischemia and reduces cerebral ischemic damage (Nogawa et al., 1997). Thus, COX-2 reaction products may contribute to neuronal death in peripheral regions of the ischemic territory at risk for infarction. The mechanisms of COX-2 neurotoxicity are likely to include synthesis of superoxide anions and pro-inflammatory prostanoids (Wu, 1995).
Recent evidence indicates that after cerebral ischemia there is an interaction between iNOS-derived NO and COX-2. Although iNOS-positive neutrophils are in close spatial proximity to COX-2-positive neurons, inhibition of iNOS by aminoguanidine attenuates COX-2 reaction products only in the ischemic region (Nogawa et al., 1998). Furthermore, postischemic prostaglandin production is reduced in iNOS null mice despite normal COX-2 mRNA and protein expression (Nogawa et al., 1998). These observations indicate that, in the ischemic brain, NO produced by iNOS drives COX-2 activity and increases its catalytic output.
The interaction between iNOS-derived NO and COX-2 raises the possibility that some of the deleterious effects of ischemia-induced iNOS expression are mediated through COX-2 reaction products. In this study, we sought to explore this possibility by assessing the relative contribution of iNOS and COX-2 to ischemic brain damage. We reasoned that if the pathogenic effects of iNOS-derived NO and COX-2 are mediated through largely independent mechanisms, then inhibition of COX-2 in iNOS null mice (iNOS −/−) should confer a protection greater than that attained by iNOS deletion alone. On the other hand, if iNOS-derived NO mediates some of its deleterious effects through COX-2, then inhibition of COX-2 in iNOS −/− mice should not provide added protection. We found that the COX-2 inhibitor NS398 attenuates ischemic brain damage in wild-type mice but not in iNOS −/− mice. The data support the hypothesis that COX-2 activation is an important pathogenic factor in the damage produced by iNOS-derived NO.
METHODS
Animals
Studies were approved by the Animal Care and Use Committee of the University of Minnesota. Experiments were conducted in 14 C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME), 11 129/SVeV (SV129) mice (Jackson Laboratories) and 27 iNOS −/− mice. All mice were males. As described in detail elsewhere (Iadecola et al., 1997), iNOS −/− mice were obtained from a colony established from breeding pairs provided by Dr. C. Nathan and J. Mudgett (MacMicking et al., 1995). Mice were studied at age 8 to 9 weeks.
Middle cerebral artery occlusion
Focal cerebral ischemia was produced by occlusion of the MCA as previously described (Iadecola et al., 1997; Zhang et al., 1997). Briefly, mice were anesthetized with halothane (induction 5%, maintenance 1 %) in an oxygen-nitrogen mixture.
Body temperature was maintained at 37 ± 0.5 °C by a thermostatically controlled infrared lamp. A 2-mm hole was drilled at a site superior and lateral to the left foramen ovale to expose the left MCA. The MCA was elevated and cauterized distal to the origin of the lenticulostriate branches. Wounds were sutured, and mice were returned to their cages and closely monitored until they recovered from anesthesia. Rectal temperature was measured and controlled until mice regained full consciousness. Thereafter, rectal temperature was measured at 24-hour intervals (before and 2 hours after each NS398 administration) until the time of they were killed. All mice survived until they were killed for determination of infarct volume.
Determination of infarct volume
Mice were killed for measurement of infarct volume 96 hours after MCA occlusion, shortly after the last neurologic evaluation. As described in detail elsewhere (Iadecola et al., 1997; Zhang et al., 1997), brains were removed and frozen in cooled isopentane (−30°C). Coronal forebrain sections (thickness, 30 μm) were serially cut in a cryostat, collected at 150 μm intervals, and stained with thionin. Infarct volume was determined by an image analyzer (MCID, Imaging Research Inc.) (Iadecola et al., 1997; Zhang et al., 1997). To factor out the contribution of ischemic swelling to the volume of the lesion, infarct volume in cerebral neocortex was corrected for swelling using the procedure developed by Lin et al. (Iadecola et al., 1997; Lin et al., 1993; Zhang et al., 1997).
Neurologic evaluation
Neurologic deficits were assessed by a neurologic scoring system using a modification of the postural reflex test, as previously described (Bederson et al., 1986; Iadecola et al., 1997). The examiner was not aware of the identity of the mice. Neurologic scores were: 0, normal motor function; 1, flexion of torso and contralateral forelimb when mouse was lifted by the tail; 2, circling to the contralateral side when mouse was held by the tail on a flat surface, but normal posture at rest; 3, leaning to the contralateral side at rest; 4, no spontaneous motor activity. Mice were evaluated before MCA occlusion and at 24-hour intervals up to 96 hours after MCA occlusion.
Monitoring of CBF
Techniques used for monitoring CBF in anesthetized mice have been described in detail previously (Iadecola et al., 1997). Mice were anesthetized with halothane (maintenance 1 %), and the femoral artery and trachea were cannulated. Mice were artificially ventilated with an oxygen-nitrogen mixture by a mechanical ventilator (SAR-830, CWI Inc., Ardmore, PA, U.S.A.). The oxygen concentration in the mixture was adjusted to maintain arterial PO2 between 120 and 150 mm Hg. End-tidal co2 was continuously monitored using a co2 analyzer (Capstar-100, CWI Inc.) and maintained at 2.6% to 2.7% which corresponds to a Pco2 of 33 to 35 mm Hg (Iadecola et al., 1997). The physiologic parameters of the mice studied were: mean arterial pressure: 95 ± 8 mm Hg; Pco2: 33.4 ± 1.5 mm Hg; Po2: 139 ± 15 mm Hg; pH: 7.42 ± 0.03. CBF was monitored by a laser-Doppler flow probe (Vasamedic, St. Paul, MN, U.S.A.) placed through a burr hole drilled over the parietal cortex. The zero value for CBF was obtained after the mice were killed and was used to calculate percent changes in CBF (Iadecola et al., 1997).
Experimental protocol
Effect of NS-398 on infarct volume. C57BL/6, 129/SVeV, and iNOS null mice were studied. The COX-2 inhibitor NS398 (20 mg/kg; intraperitoneally) or vehicle was administered twice daily starting 6 hours after MCA occlusion. NS398 was diluted in distilled water at pH 10. Vehicle consisted of distilled water only (pH = 10). The dose of NS398 used was found in previous experiments to inhibit postischemic prostaglandin accumulation (Nogawa et al., 1998; Nogawa et al., 1997). NS398 does not affect NOS activity (Zingarelli et al., 1997). Rectal temperature was monitored daily. Mice were killed 96 hours after MCA occlusion, and brains were processed for measurement of infarct volume.
Effect of 7-NI on infarct volume in iNOS −/− mice. The relatively selective neuronal NOS (nNOS) inhibitor 7-nitroindazole (7-NI; 50 mg/kg; n = 6) or vehicle (oil; n = 5) was administered intraperitoneally 5 minutes after MCA occlusion as previously reported (Kamii et al., 1996). This dose of 7-NI inhibits forebrain NOS activity by 74% in rodents (Iadecola and Zhang, 1996). Mice were killed 96 hours after MCA occlusion for determination of infarct volume.
Effect of NS398 on CBF in iNOS −/− mice. In these experiments, the effect of NS398 on resting CBF and on the vasodilation produced by hypercapnia were studied in iNOS −/− (n = 6). Hypercapnia was induced by introducing CO2 in the circuit of the ventilator (Iadecola et al., 1997). First, resting CBF and the increase in CBF produced by hypercapnia (pCo2: 50 to 60 mm Hg) were established. Then, NS398 was administered (20 mg/kg; intraperitoneally) and effects on resting CBF and hypercapnic vasodilation studied 30 to 45 minutes later.
Data analysis
Data in text, table,.and figures are expressed as mean ± SD. Comparisons among multiple groups were statistically evaluated by one-way analysis of variance followed by post hoc Tukey-Kramer honestly significant difference test (Systat, Evanston, IL, U.S.A.). Two-group comparisons were analyzed by the paired or unpaired two-tailed t test as appropriate. Neurologic scores were evaluated by nonparametric statistical procedures. Two-group comparisons were analyzed by the Mann-Whitney U analysis (Kirk, 1982), whereas multiple comparisons were analyzed by the Kruskal-Wallis test followed by post hoc Tukey-Kramer honestly significant different test (Kirk, 1982). For all statistical procedures, differences were considered significant at P < .05.
RESULTS
Effect of NS398 on infarct volume in C57BL/6 mice
Before MCA occlusion, rectal temperature was 35.7 ± 0.9 °C in vehicle-treated mice (n = 7) and 36.0 ± 0.7 °C in mice treated with NS398 (n = 7). Treatment with NS398 did not alter rectal temperature during the 4-day survival period (P > .05; analysis of variance). For example, 24 hours after MCA occlusion, rectal temperature was 36.0 ± 0.7° C before NS398 and 36.2 ± 0.3 2 hours after. In vehicle-treated mice, MCA occlusion produced an infarct involving mostly the cerebral cortex (Table 1). Size and regional distribution of the infarct were similar to those previously reported (Iadecola et al., 1997; Zhang et al., 1997). Administration of NS398 reduced infarct volume (P < .05; t-test; Table 1) and ameliorated the motor deficits produced by MCA occlusion (P < .05 at 96 hours; Mann-Whitney U test)(Fig. 1).
Effect of NS398 on infarct volume after middle cerebral artery occlusion in wild-type mice
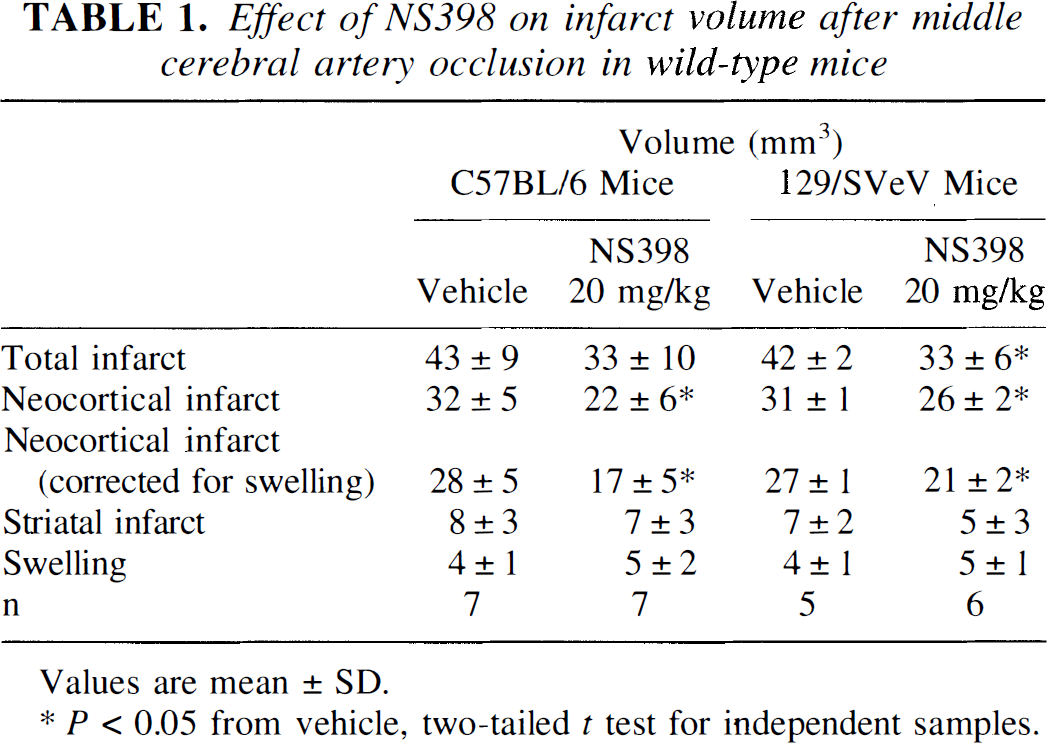
Values are mean ± SD.
P < 0.05 from vehicle, two-tailed t test for independent samples.
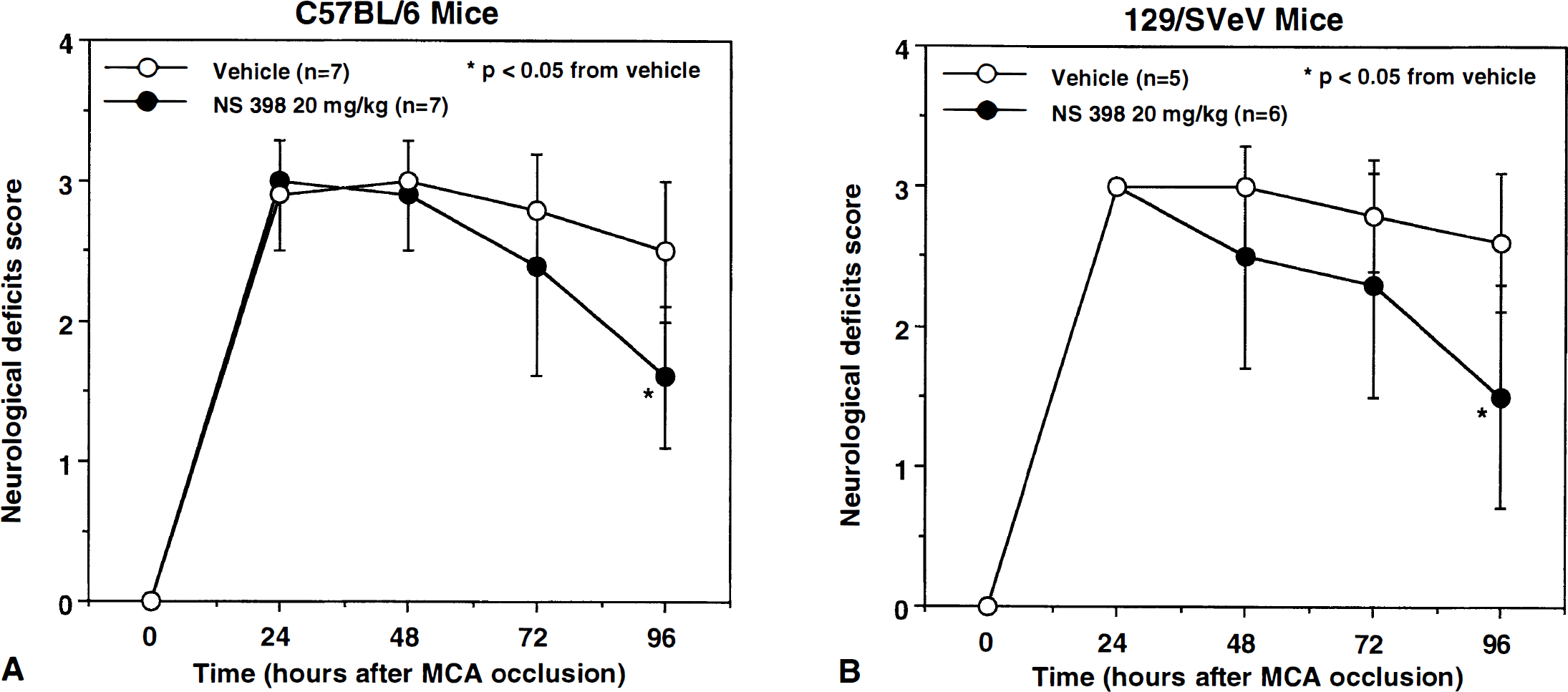
Effect of the cyclooxygenase-2 inhibitor NS398 on the motor deficits produced by middle cerebral artery occlusion. Administration of NS398 reduced the neurologic deficits both in C57BU6
Effect of NS398 or 7-NI on infarct volume in iNOS −/− mice
Before MCA occlusion, rectal temperature was 36.6 ± 0.7 °C in vehicle-treated iNOS −/− mice (n = 5) and 36.1 ± 0.8 °C in mice treated with NS398 (n = 5). Treatment with NS398 did not alter rectal temperature during the 4-day survival period (P > .05; analysis of variance). In agreement with previous observations, infarct volume in iNOS −/− mice was smaller than that of C57BL/6 mice (−32 ± 14% in neocortex; P < .05) (cf. Iadecola et al., 1997). However, in contrast to C57BL/6 mice, treatment with NS398 did not reduce infarct volume (Table 2). Rather, NS398 tended to increase the volume of injury, although the effect did not reach statistical significance (P > .05). NS398 did not affect the motor deficits produced by MCA occlusion (P> .05; Fig. 2). To rule out the possibility that the lack of effectiveness of NS398 in iNOS −/− mice was due to the fact that infarct volume was already maximally reduced, we studied the effect of the nNOS inhibitor 7-NI on infarct volume in iNOS −/− mice. Treatment with 7-NI (n = 6) reduced neocortical infarct volume by 20 ± 10% compared to vehicle-treated controls (P < .05; n = 5).
Effect of NS398 on infarct volume after middle cerebral artery occlusion in iNOS −/− mice
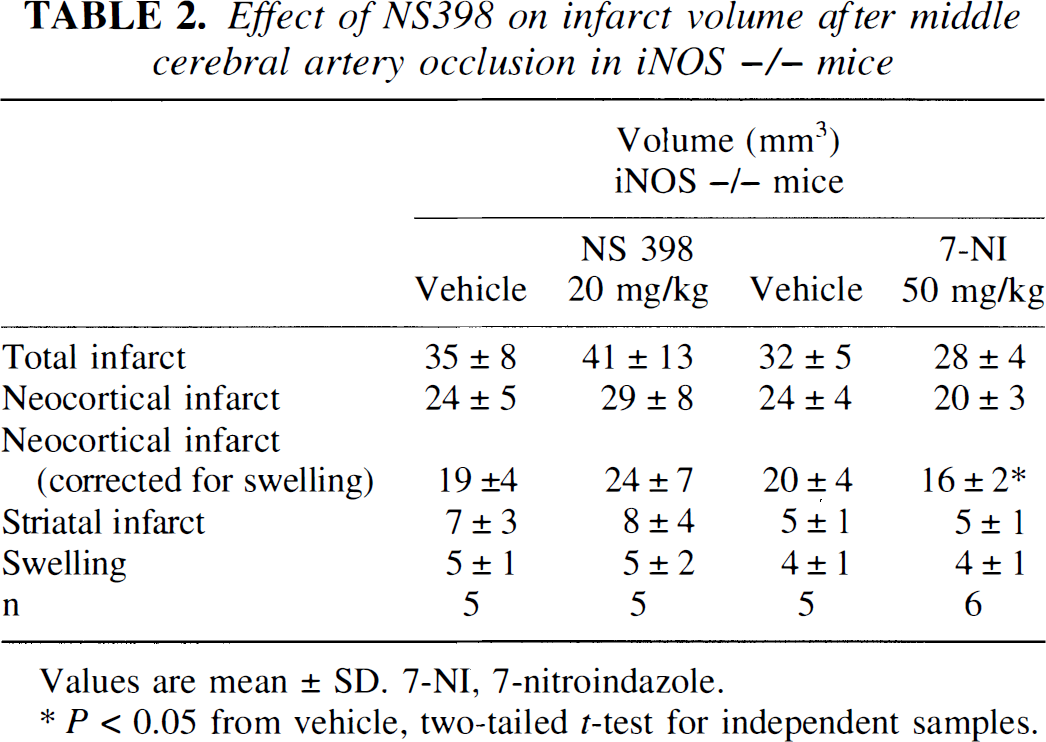
Values are mean ± SD. 7-NI, 7-nitroindazole.
p < 0.05 from vehicle, two-tailed t-test for independent samples.
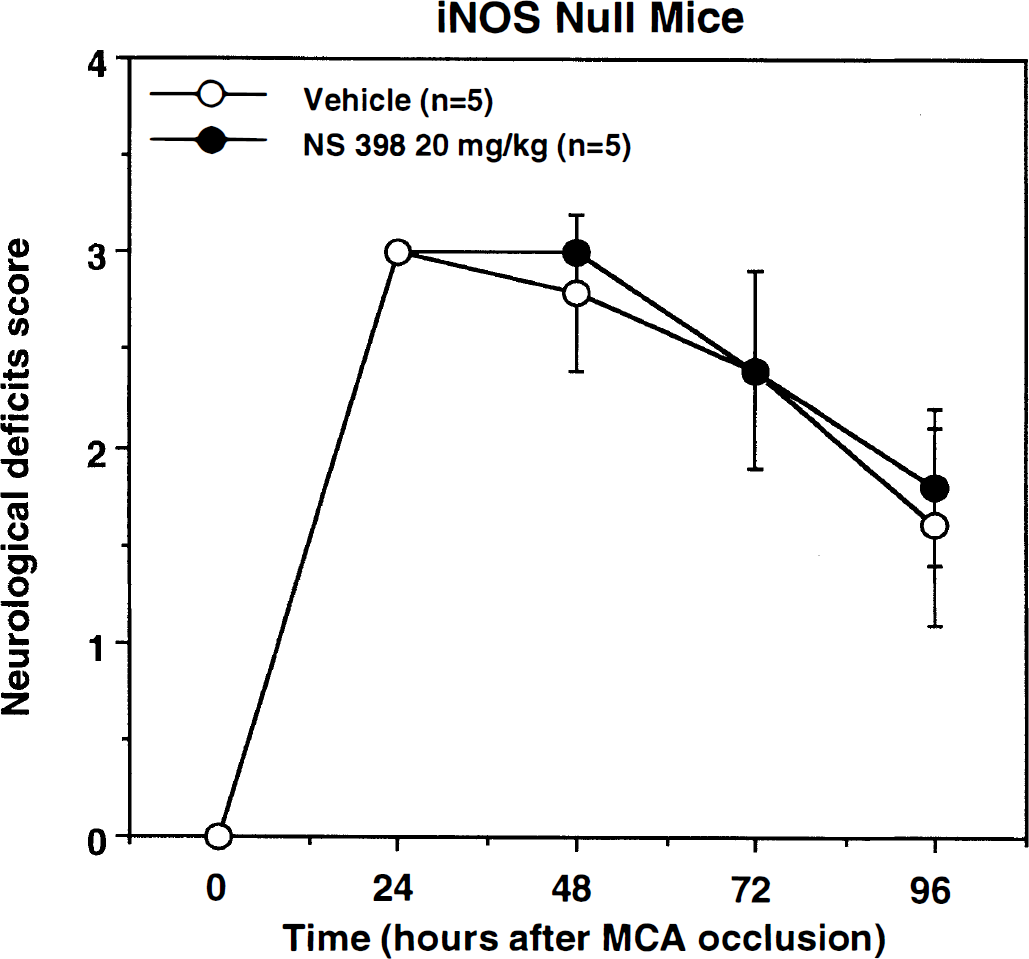
Effect of the cyclooxygenase-2 inhibitor NS398 on the motor deficits produced by middle cerebral artery occlusion in iNOS −/− mice. At variance with C57BL/6 and 129/SVeV mice (cf. Fig. 1). administration of NS398 did not reduce the neurologic deficits in iNOS −/− mice (P < .05; Mann-Whitney U test).
Effect of NS398 on infarct volume in 129/SVeV mice
iNOS −/− mice, as most mice generated by homologous recombination, have a mixed genetic background comprising both C57BL/6 and 129/SVeV strains (MacMicking et al., 1995). To rule out the possibility that the lack of protection by NS398 in iNOS −/− mice was related to strain differences, we tested the effect of NS398 on infarct volume in 129/SVeV mice. In vehicle-treated mice (n = 5), infarct volume did not differ from that of C57BL/6 mice (Table 1; P > .05). Treatment with NS398 (n = 6) reduced neocortical infarct volume by 22 ± 7% (Table 1; P < .05) and ameliorated the motor deficits produced by MCA occlusion (P < .05 at 96 hours) (Fig. 1).
Effect of NS398 on resting CBF and on the reactivity to hypercapnia in iNOS −/− mice
In these experiments (n = 6), we sought to explore the possibility, that the absence of protection by NS398 in iNOS −/− mice was related to cerebral hemodynamic effects of NS398 present only in iNOS −/−. Mean arterial pressure was 95 ± 8 mm Hg before NS398 administration and 90 ± 7 60 minutes after (P > .05). NS398 did not affect resting CBF or the CBF increase produced by hypercapnia (Fig. 3). The intensity of the hypercapnia challenge was comparable before and after NS398 (before: Pco2 = 54.5 ± 3 mm Hg; after: 55.3 ± 2; P > .05).
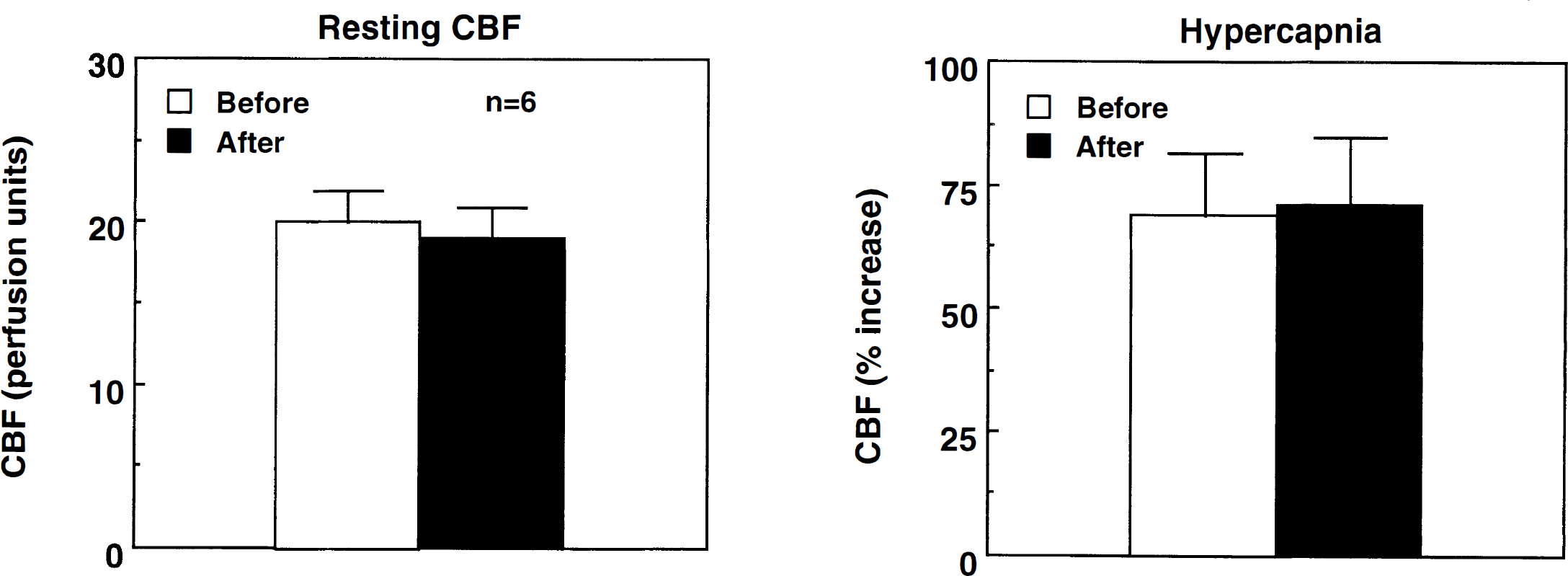
Effect of NS398 on resting CBF
DISCUSSION
We sought to investigate further the role of iNOS and COX-2 in focal cerebral ischemic damage. Previous investigations have indicated that NO produced by iNOS increases COX-2 catalytic output, raising the possibility that COX-2 reaction products contribute to the brain damage produced by iNOS-derived NO (Nogawa et al., 1998). In this study, we sought to provide evidence in support of this hypothesis. We reasoned that if iNOS-derived NO is required for the toxic effects of COX-2 to take place, then inhibition of COX-2 in iNOS −/− mice, which do not express iNOS after cerebral ischemia (Iadecola et al., 1997), should not provide added protection. Consistent with this prediction we found that the COX-2 inhibitor NS398 reduced the infarct produced by MCA occlusion in wild-type mice but not in iNOS −/−mice. This observation indicates that iNOS-derived NO is required for the toxicity of COX-2 reaction products to occur.
The lack of effectiveness of NS398 in iNOS −/− mice cannot be attributed to genetic differences in the susceptibility to this inhibitor in the parental strains because NS398 reduced ischemic damage both in C57BL/6 and in 129/SVeV mice, the two strains used to generate the null mice (MacMicking et al., 1995). Similarly, the absence of protection cannot result from the fact that iNOS −/− mice are maximally protected from cerebral ischemic damage because administration of the nNOS inhibitor 7-NI reduced infarct volume in iNOS −/− mice. Furthermore, the failure of NS398 to reduce infarct volume in the iNOS −/− mice cannot be attributed to confounding cerebrovascular effects of NS398. (1) NS398 was administered 6 hours after induction of cerebral ischemia, at a time when hemodynamic effects are no longer able to influence the outcome of permanent cerebral ischemia (e.g., Zhang and Iadecola, 1994). (2) Administration of NS398, at the same dose used in ischemia experiments, did not alter arterial pressure, resting CBF, or the reactivity of CBF to hypercapnia in iNOS null mice. While these observations are preliminary and need to be expanded to other aspects of cerebrovascular regulation, they suggest that NS398 is devoid of cerebrovascular effects that could potentially antagonize the protection exerted by the drug. Therefore, the lack of protection by NS398 in iNOS −/− mice is unlikely to be caused by factors other than deletion of iNOS gene.
The mechanisms by which NO and its derived chemical species exert their cytotoxic effect are multiple and are thought to include inhibition of ATP-producing enzymes (Kroncke et al., 1997), DNA damage (Szabo and Ohshima, 1997), and oxidative damage produced by peroxynitrite, a highly reactive chemical species formed by the reaction of NO with superoxide (Beckman et al., 1990). Peroxynitrite-induced DNA single-strand breaks activate the DNA repair enzyme poly(ADP)ribose synthetase which worsens energy depletion and contributes to ischemic brain injury (Elias son et al., 1997; Szabo and Dawson, 1998). In addition, DNA strand breaks induce the expression of p53, a tumor-suppressor protein that induces cell cycle arrest and apoptosis (Forrester et al., 1996). The results of the present study suggest that iNOS-derived NO could produce tissue damage also by activating COX-2 and increasing the toxic output of this enzyme. Therefore, COX-2 reaction products could be an additional mechanism by which NO exerts its deleterious effects on the postischemic brain.
However, the mechanisms by which COX-2 contributes to cerebral ischemic injury have not been elucidated. One possibility is that pro-inflammatory prostanoids synthesized by COX-2 contribute to the inflammatory reaction that involves the ischemic brain and, by this mechanism, worsen damage (Seibert et al., 1995). In addition, COX-2 could act as a source of superoxide for peroxynitrite formation. The two catalytic steps of COX-2, cyclooxygenase and peroxidase, are associated with production of reactive oxygen species (Vane et al., 1998). Superoxide produced by COX-2 activity could react with NO to form peroxynitrite, a highly reactive oxidant thought to mediate many of the toxic effects of NO (Kroncke et al., 1997). Irrespective of its mechanisms, the present results suggest that iNOS-derived NO is required for the full expression of COX-2 neurotoxicity.
Another new finding of this study is that NS398 reduces ischemic brain damage also in mice. The reduction in infarct volume afforded by NS398 cannot be attributed to effects on body temperature, arterial pressure or blood gases. While NS398 does not influence rectal temperature (present study), this inhibitor does not affect arterial pressure in anesthetized mice (present study) or in awake rats (Nogawa et al., 1997). Because of their small blood volume, serial measurements of arterial blood gases were not made in the mice in which infarct volume was studied. However, experiments in rats indicate that NS398 does not affect blood gases (Nogawa et al., 1997). Selective COX-2 inhibitors have previously been reported to reduce the damage resulting from focal ischemia or global ischemia in rats (Nakayama et al., 1998; Nogawa et al., 1997). The observation that the COX-2 inhibitor NS398 reduces ischemic damage also in mice suggests that the participation of COX-2 in ischemic brain injury is preserved across species. However, it would be important to determine whether COX-2 inhibition reduces infarct volumes also in other stroke models, such as those with reperfusion. We have recently found that iNOS and COX-2 are expressed also in the human brain in the acute stages of focal cerebral ischemia (Forster et al., 1999; Iadecola et al., 1999). As in rodents, iNOS expression occurs in neutrophils and vascular cells whereas COX-2 expression occurs also in neurons at the infarct border. While it remains to be established whether iNOS-derived NO and COX-2 reaction products contribute to the pathogenesis of human stroke, the similarity in the timing and cellular localization of iNOS and COX-2 between rodents and humans suggests similar roles in the mechanisms of damage. nNOS inhibition reduces infarct volume in iNOS null mice. This observation confirms that both nNOS and iNOS contribute to ischemic brain injury (Iadecola, 1997). However, the fact that nNOS inhibition in iNOS null mice does not completely eliminate ischemic damage underscores the pathogenic importance of factors independent of the NO pathways.
In conclusion, we have shown that the COX-2 inhibitor NS398 reduces cerebral ischemic damage in wild-type mice but not in iNOS −/− mice. The effect cannot be attributed to variations in the genetic background of iNOS −/− mice, to cerebral hemodynamic effects of NS398, or to the fact that the maximal protection possible was achieved in the iNOS −/− mice. The data suggest that iNOS-derived NO is required for the expression of toxicity mediated by COX-2 reaction products. Thus, COX-2 reaction products may be an important factor in the deleterious effect exerted by NO derived from iNOS in the postischemic brain.
Footnotes
Acknowledgments:
The authors thank Ms. Deborah Kabes for editorial assistance and Ms. Tracy Aber for managing the mouse colony.