
Abstract
In this study we show that the aminopeptidase N of cerebral pericytes (pAPN) associated with the blood—brain barrier (BBB) is downregulated in pericytic cell cultures. This observation is in accordance with previous data describing comparable in vitro effects for BBB-specific enzymes of endothelial or pericytic origin, such as γ-glutamyl transpeptidase or alkaline phosphatase. By polymerase chain reaction and in situ hybridization we were able to determine that the downregulation of pAPN occurs at the posttranscriptional level. The mRNA of pAPN was found to be constitutively expressed even when the protein is no longer detectable. Culturing the pericytes in an endothelial cell—conditioned medium allowed pAPN to be reexpressed. However, the reexpression effect depended largely on the culturing conditions of the pericytes. Although purified pericytes deprived of endothelial cells did not reveal a reexpression effect, pericytes that were kept in contact with endothelial cells were able to acquire a pAPN-positive phenotype, indicating that endothelial cells constitute an essential requirement for the in vitro reexpression of pAPN. Astrocytes, however, were insufficient in exerting any reexpression effect.
The blood—brain barrier (BBB) regulates the exchange of blood solutes with the interstitial cerebral fluid and is composed of a physical and metabolic barrier. The BBB consists of a microvascular endothelium with its cellular adjuncts (Reese and Karnovsky, 1967; Dermietzel, 1975; Crone and Levitt, 1984; Dermietzel and Krause, 1991). In recent years considerable efforts have been focused on models that mimic BBB conditions in vitro (Audus and Borchardt, 1987; Greenwood, 1991; Rubin et al., 1991; Joo, 1992; Abbott et al., 1992). The results of these experiments have strengthened the essential role of endothelial cells for BBB mechanisms. They have also provided evidence that endothelium-associated cells, like astrocytes and pericytes, are required for the establishment and maintenance of intrinsic BBB functions (Farrell et al., 1987; Maxwell et al., 1987, 1989; Meyer et al., 1990, 1991; Frey et al., 1991; Risau et al., 1992).
Astrocytes, which tightly clasp cerebral microvessels by their endfeet, have been demonstrated to exhibit inductive capabilities on the formation and functional efficiency with respect to the tightness of the cerebral endothelium (Janzer and Raff, 1987), the most prominent physiologic feature of the intact BBB (Reese and Karnovsky, 1967; Stewart and Wiley, 1981; Janzer and Raff, 1987; Rutten et al., 1987). Furthermore, the expression of metabolic components of the BBB is suggested to be governed by regulatory influences provided by astrocytes. Prominent metabolic components of the BBB are the glucose transporter(s) (Glut-1) (Maxwell et al., 1989), alkaline phosphatase (Meyer et al., 1991; Roux et al., 1994), γ-glutamyl transpeptidase (Maxwell et al., 1987; Meyer et al., 1991; Mizuguchi et al., 1994; Roux et al., 1994), the low-density lipoprotein-receptor (Dehouck et al., 1994), the transferrin transporter system (Roberts et al., 1993), acetylcholinesterase (Pakaski and Kasa, 1992), and the Na-K-Cl-cotransporter (Sun et al., 1995). Recent findings, however, have led to a reconsideration of the role that astrocytes play in the process of BBB induction. For instance, the studies of Holash et al. (1993) substantiate the general concept that astrocytes exhibit significant effects that maintain or influence functional properties of the BBB, but they lack the ability to initially induce BBB formation. The simplistic view of a bilateral axis in BBB induction and maintenance involving exclusively astrocytes and endothelial cells has become debatable (see Dermietzel and Krause, 1991).
Another cell type coming into focus in BBB research is the cerebral pericyte, a morphologic, biochemical, and physiologic heterogeneous cell population. Pericytes, which share a common basement membrane with the cerebral endothelium, are suggested to have phagocytotic potency (Cancilla et al., 1972; Jordan and Thomas, 1988), which operates as a “second line of defense” at the boundary between blood and brain (Farrell et al., 1987). Furthermore, pericytes are considered to be influential in the regulation of capillary blood flow (Le Beux and Willemot, 1980; Joyce et al., 1985a, 1985b; Herman and D' Amore, 1985). In peripheral tissues pericytes have been reported to provide (1) potential modulators of endothelial permeability (Imayama and Urabe, 1984), (2) stabilizing effects on microvessel walls (Nehls and Drenckhahn, 1993), and (3) promoting activity on angiogenic processes (Orlidge and D' Amore, 1987; Klagsbrun and D' Amore, 1991) and capillary sprouting (Verhoeven and Buyssens, 1988; Nehls et al., 1992, 1994). Evidence is also accruing that cerebral pericytes constitute a major component of the metabolic BBB by expressing specific enzymes, such as γ-glutamyl transpeptidase (Frey et al., 1991; Risau et al., 1992), or enzymes involved in neurotransmitter metabolism, like glutamyl aminopeptidase (Bausback et al., 1988; Healy and Wilk, 1993; Song et al., 1993).
Consistent with these observations, we recently reported on the expression of a specific isoform of aminopeptidase N (CD 13) in cerebral pericytes (pAPN) (Krause et al., 1988; Kunz et al., 1994). The lack of pAPN expression in brain regions devoid of a tight endothelium, like the circumventricular organs, suggested that pAPN is a member of the metabolic complement of the BBB that is involved in neuropeptide degradation (Mizutani et al., 1993), i.e., APN has been suggested to be involved in the degradation of endogenous endorphins, like enkephalins (De la Baume et al., 1983). We also acquired evidence that pAPN responds to pathologic disturbances of the BBB (Kunz et al., 1995).
In essence, cerebral pericytes are an important member of the cellular complex that constitutes the BBB. However, detailed data on the cell biology of this cell class of the BBB are still lacking.
To gain a better insight into the cellular biologic properties of cerebral pericytes, we designed a method to purify pericytes of brain microvessels. By exploiting pAPN as a specific marker for cerebral pericytes, we found that (1) purified cerebral pericytes are unable to preserve their brain-specific phenotype in culture, as shown by loss of pAPN expression, (2) downregulation of pAPN occurs at the translational level, (3) (re)induction of pAPN can only be achieved in coculture with endothelial cells, (4) cultured primary astrocytes are insufficient to (re)induce the pericytic phenotype, and (5) (re)induction requires a soluble factor (or factors) provided by endothelial cells.
MATERIALS AND METHODS
Antibodies and immunologic techniques
A number of monoclonal and polyclonal antibodies were applied for the characterization of cell specificity under immunofluorescence conditions. The following antibodies were applied:
Monoclonal antibodies. The antibody directed against pAPN was generated in our laboratory and has been extensively characterized (Krause et al., 1988; Kunz et al., 1994, 1995). Undiluted hybridoma supernatants were used for immunocytochemical detection as well as for solid-phase isolation (see below). Anti-EDI (MCA 341), an antibody recognizing reactive cells of monocytic origin (macrophages) was purchased from Serotec (Wiesbaden, Germany) and applied in a dilution of 1:250. The anti-MUC 102 antibody, which is regarded to be specific for microglial cells (Gehrmann and Kreutzberg, 1991), was kindly provided by G. Kreutzberg (Max-Planck Institute of Psychiatry, Munich, Germany). The working solution of this monoclonal antibodies (mAb) was 1:700. Monoclonal antibodies against smooth muscle (sm)-actin and vimentin were purchased from Sigma (Munich, Germany) and Boehringer (Mannheim, Germany), respectively. The applied dilutions were 1:400 for anti-sm actin and 1:150 for anti-vimentin.
Polyclonal antibodies. For recognition of cerebral endothelial cells an antibody directed against the BBB-specific Glut-1 (dilution 1:50) was used. The anti-Glut-1 antibody was prepared and affinity-purified as described earlier (Wang, 1987; Dermietzel et al., 1992). An antibody against the factor VIII-related antigen (Dakopats, Hamburg, Germany) was applied in a dilution of 1:1,000 and served as a general marker for endothelial cells. Primary astrocytes were identified by the expression of glial fibrillary acidic protein (GFAP) by means of an anti-GFAP antibody (dilution 1:200; Sigma).
Fluorescein isothiocyanate- (FITC) and Texas red—coupled secondary antibodies (Sigma) were used in a dilution of 1:400.
Immunocytochemistry. For immunologic detection of cell-specific proteins, indirect immunocytochemistry was performed as described earlier (Kunz et al., 1994). In brief, cells were washed twice with 0.1 mol/L phosphate-buffered saline (PBS) to remove culture medium, followed by a brief fixation in absolute ethanol for 10 minutes at room temperature. After several brief washes with 0.1 mol/L PBS, nonspecific binding sites were saturated by incubation of the cells in 0.1 mol/L PBS containing 0.1% bovine serum albumin (BSA) and 10% horse serum as additives. Blocking time was 30 minutes. For double immunostaining, cells were sequentially incubated with a monoclonal and a polyclonal primary antibody for 60 minutes each at room temperature, followed by thorough rinsing with 0.1 mol/L PBS with 0.1% BSA, and subsequent incubation with the FITC- or Texas red-coupled secondary antibodies, or both, in the dark for 20 minutes each at room temperature. After three additional washes with 0.1 mol/L PBS with 0.1% BSA, specimens were embedded in FITC-Guard (Testog, Chicago, IL, U.S.A.) and subjected to immunofluorescence microscopy on an Axiovert 35 microscope (Zeiss, Oberkochen, Germany).
Isolation of cells and cell culture procedures
Cerebral microvascular cells were isolated as described earlier (Kunz et al., 1994). In brief, cells were obtained from the cerebral cortex of 8 to 10 rat brains (Wistar strain, 8 to 12 weeks old) by collagenase digestion (collagenase type CLS II; Seromed, Berlin, Germany) followed by a subsequent 50% Percollgradient (Pharmacia, Freiburg, Germany), and centrifuged at 1,250g (4°C) for 10 minutes (Risau et al., 1992). The collected microvascular cells were washed twice with 0.1 mol/L PBS. The isolated cells were then used for cell culture as crude microvascular cell fraction (CMF) or further subjected to an additional purification step to isolate purified cerebral pericytes (see below). Samples of the CMF were seeded on poly-
Purification of cerebral pericytes was achieved by a solid-phase technique making use of immunomagnetic beads. For purification microvascular cells were prepared as described above. The collected fraction was then resuspended in 50 µL DMEM supplemented with 4.5% glucose. Dissociation was achieved with a dissociation solution (Sigma), used in a concentration of 4 mL per 50 µL of cell suspension, by repeated tituration with a glas pipette at room temperature. After centrifugation (160g, 5 minutes), samples were incubated with 500 µL of the undiluted supernatant of anti-pAPN antibody for 30 minutes at room temperature, washed twice with DMEM with 4.5% glucose, and finally resuspended in another 500 µL DMEM with 4.5% glucose. A Dynabead M-450 solution (Dynal, Oslo, Norway) coated with goat anti-mouse IgG was added, achieving a total concentration of 107 beads/mL cell suspension. After incubation for 45 minutes at 4°C under constant shaking, samples were placed in a Dynal magnet (MPC; Dynal) to separate immunoadsorbed cells. Separation time was 2 minutes. Supernatants containing free microvascular cells not immunoadsorbed to the Dynabeads were soaked off and further used as mixed culture samples (MCS). The purified pericytes were collected, washed twice with DMEM with 4.5% glucose, and seeded into plastic culture dishes (25 cm2). Both the purified pericyte cultures (PPC) and MCS, which consist chiefly of microvascular fragments and free unbound pericytes, were grown under the same culture conditions as described above for CMF.
Cerebral astrocytes were isolated from cerebral cortices of 1-to 2-day-old rats (Wistar strain) for coculture with PPC and MCS as described previously (Dermietzel et al., 1991). To achieve highest purity of astrocytic cell cultures (>99%), contaminating macrophages and oligodendrocytes were separated by a series of continuous shaking for 6, 18, and 24 hours. Contaminating cells were removed by changing the culture medium after each agitation step. For coculture experiments first passage astrocytes were used.
A cerebral endothelial cell line bEnd.3 (Montesano et al., 1990; kindly provided by D. Männel, University of Regensburg) was further used for coculture experiments. The bEnd.3 cells were cultured at 37°C (5% CO2) in DMEM containing 4.5% glucose and 5% fetal calf serum in plastic culture flasks. After acquiring confluency (1 to 2 days after seeding), bEnd.3 cells were passaged for coculturing with microvascular cell fragments or purified pericytes.
Coculture conditions. Primary astrocytes and bEnd.3 were subjected to coculture with PPC and MCS.
Coculturing was performed under the following conditions. First, conditions with unrestricted physical contact were achieved by seeding PPC or MCS on a subconfluent layer of astrocytes (alternatively, bEnd.3 cells). Second, culture conditions with limited cell-cell contact were realized using perforated polycarbonate membranes (pore size approximately 1 µm; Becton-Dickinson, Lincoln Park, NJ, U.S.A.). Membranes were loaded on one side with the feeder layer (astrocytes or bEnd.3 cells, respectively), which was then cultured to confluency, subsequently turned around and further loaded with purified pericytes or MCS. Membranes were fixed in a special sheath-holder device (Minucells; Minuth et al., 1992) throughout the experiments. Third, cocultures with exclusion of physical contact were obtained by cultivation of cells on both sides of glass coverslips in the same sheath-holder device.
Preparation of cerebral microvessels for Western blotting. To achieve sufficient amounts of pAPN from cerebral microvessels for Western blotting vessels were isolated as described by Mrsulja et al. (1976). This protocol achieves a higher concentration of pAPN than the Percoll method described above. All steps of isolation were performed at 4°C. In brief, cerebrum of one adult rat (Wistar strain) was homogenized in 20 volumes of Ringer's solution with 1% BSA and 10 mmol/L HEPES, pH 7.4, in a glass homogenizer with a tight-fitting pestle (10 strokes for each sample). After homogenization, the samples were centrifuged at 1,500g for 15 minutes. The pellets were resuspended in Ringer's solution with 1% BSA and centrifuged for another 10 minutes at 1,500g. The collected pellets were suspended in 10 mL of 0.25 mol/L sucrose solution, layered over a two-step discontinuous sucrose gradient of 1.0 mol/L and 1.75 mol/L sucrose (wt/vol; 12 mL each), and centrifuged at 58,000g for 30 minutes in a Beckman ultracentrifuge (rotor SW-28). The interphase between the 1.0 mol/L and 1.75 mol/L sucrose was collected and checked for microvessel purity by phase-contrast microscopy.
Biochemical and molecular biologic techniques
Preparation of kidney brush-border membranes. Preparation of the brush-border membranes from rat kidney proximal tubules was done as described by Biber et al. (1981). All steps were performed at 4°C. Cortices of two kidneys from one adult rat (Wistar strain, 3 to 6 months old) was homogenized and centrifuged for 15 minutes at 1,800g. The supernatant was centrifuged for a further 30 minutes at 22,300g. Collection of pellets and homogenization and centrifugation steps were repeated twice, as described above. The pellets of the final centrifugation step were highly enriched in brush-border membranes.
Polyacrylamide gel electrophoresis and Western blotting. Samples of kidney brush-border membranes, homogenates of freshly isolated cerebral microvessels, and cultivated PPC were separated on a one-dimensional continuous 7.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel. Proteins were transferred to a polyvinylidene difluoride membrane (0.45 µm; Millipore, Eschborn, Germany) by the semidry blotting method. For immunoincubation, the membrane was blocked in PBS with 0.05% Tween 20 at 4°C overnight. The incubation with the monoclonal anti-pAPN antibody was performed at room temperature for 2.5 hours, followed by the secondary antibody (anti-mouse IgG antibody coupled to 10-nm gold beads; Aurion, Wageningen, the Netherlands) overnight. Bands labeled with immunogold were visualized by silver enhancement according to the recommendation of the manufacturer (Aurion).
Glycosidase digestion. Kidney brush-border membranes were digested with endoglycosidase F (Boehringer) to evaluate binding efficiency of the mAb anti-pAPN to the glycosylated and nonglycosylated forms of APN. For this purpose 5 µL of washed brush-border membranes were solubilized in 50 µL 1% SDS sample buffer. After heating the brush-border membranes at 100°C for 1 minute in 1% SDS sample buffer and removing the insoluble residue by centrifugation, the extract was adjusted to 25 µL endo F buffer (50 mmol/L potassium phosphate, 1% N-octylglycoside, 25 mmol/L EDTA, pH 6.5). After a further incubation at 100°C for 1 minute and cooling at 37°C, 250 mU endoglycosidase F were added. The reaction mixture was then incubated for 60 minutes at 37°C before gel electrophoresis. Gels of treated and untreated fractions were subjected to Western blotting (see above).
RNA isolation. Total RNA was isolated from purified pericytes and from microvascular cells (42 days in vitro, third passage, one culture flask of 25 cm2, each) according to Chomczynski and Sacci (1987) with slight modification. In brief, cell lysis was performed using TRIzol reagent (total RNA isolation reagent; GibcoBRL, Berlin, Germany). After addition of chloroform and centrifugation (15 minutes, 12,000g, 4°C), the aqueous phase containing the total RNA was aspirated and precipitated in isopropyl alcohol.
Reverse transcriptase polymerase chain reaction. Total RNA from PPC and MCS was used as template for amplifying a fragment of pAPN. Primer sets for pAPN cDNA amplification and Reverse transcriptase polymerase chain reaction (RT-PCR) conditions were identical to those described recently (Kunz et al., 1994).
Southern blotting. For Southern blotting, an agarose gel (2%) was loaded with 20 ng of each RT-PCR cDNA sample obtained from purified pericytes or MCS. After electrophoresis, the cDNA was blotted onto nitrocellulose membranes according to standard techniques (Sambrook et al., 1989). The membranes were then hybridized overnight at 42°C to a digoxygenin (DIG; Boehringer)-labeled pAPN cDNA probe (Kunz et al., 1994). Final washes were performed under high stringency conditions in 0.1× saline sodium citrate buffer with 0.1% SDS at 65°C. The hybridized DIG-labeled complex was detected by means of an anti-DIG antibody coupled to alkaline phosphatase (Boehringer).
Nonradioactive in situ hybridization. For in situ hybridization PPC grown on poly-
After hybridization, cells were washed thoroughly with 2× SSC (0.3 mol/L NaCl, 30 mmol/L citrate buffer, pH 7.0) and 0.1 × SSC at room temperature. To remove nonhybridized cRNA, specimens were incubated with 10 mmol/L Tris and 0.5 mol/L NaCl containing ribonuclease A (6 µg/mL; Boehringer) for 10 minutes at 37°C. Cells were finally rinsed three times in 0.1× SSC at 55°C and one time in 0.1× SSC at room temperature.
For immunodetection of the DIG-labeled hybridization products, specimens were washed twice in 0.1 mol/L PBS. Nonspecific binding sites were saturated with 0.1 mol/L PBS with 1% BSA, and cells were incubated for 2 hours with a polyclonal anti-DIG antibody (1:50 in 0.1 mol/L PBS with 0.2% BSA and 0.1% Triton × 100) (Boehringer). Immunostaining was performed by means of a FITC-coupled secondary antibody as described above.
RESULTS
Isolation and characterization of cerebral microvascular cells
The history of a particular cell type is considered to be of primary importance in the process of dedifferentiation or redifferentiation events under in vitro conditions (Koechlin et al., 1991). To achieve a reproducible collection of pericytes, we first characterized fractions of these cells that differed significantly in their cell composition.
Crude microvascular cell fraction. Crude microvascular cell fraction was obtained by isolating microvessels from cortical rodent brains after subjecting them to collagenase treatment (Risau et al., 1992). By applying a number of different cell-specific antibodies, the CMF was determined to consist of a collection of endothelial cells (factor VIII-related antigen-positive; Fig. 1A), pericytes (pAPN- and sm-actin-positive; Fig. 1B,C), and to a minor degree, contaminating astrocytes (GFAP-positive; Fig. 1D). Microglial cells (MUC 102-positive; Fig. 1E) and macrophages (EDI-positive; Fig. 1F) were also found in varying amounts. In addition, the CMF contained small fragments of intact microvessels that resisted collagenase digestion (not shown). When grown under supplemented culture conditions (10% BSA and 1% bovine retina extract), pericyte proliferation was favored over the other cell species. A uniform pericyte-like cell type was achieved after 14 days in culture (Swinscoe and Carlson, 1992). This cell type was characterized by intracellular fibers of sm-actin (stress fibers; see Fig. 1C) and the formation of multicellular nodules (Schor and Schor, 1986; Schor et al., 1990). Because endothelial cells are inhibited in growth by proliferating pericytes (Orlidge and D' Amore, 1987; Swinscoe and Carlson, 1992), the formation of endothelial “cobblestone-like” monolayers was rare and transient in CMF.
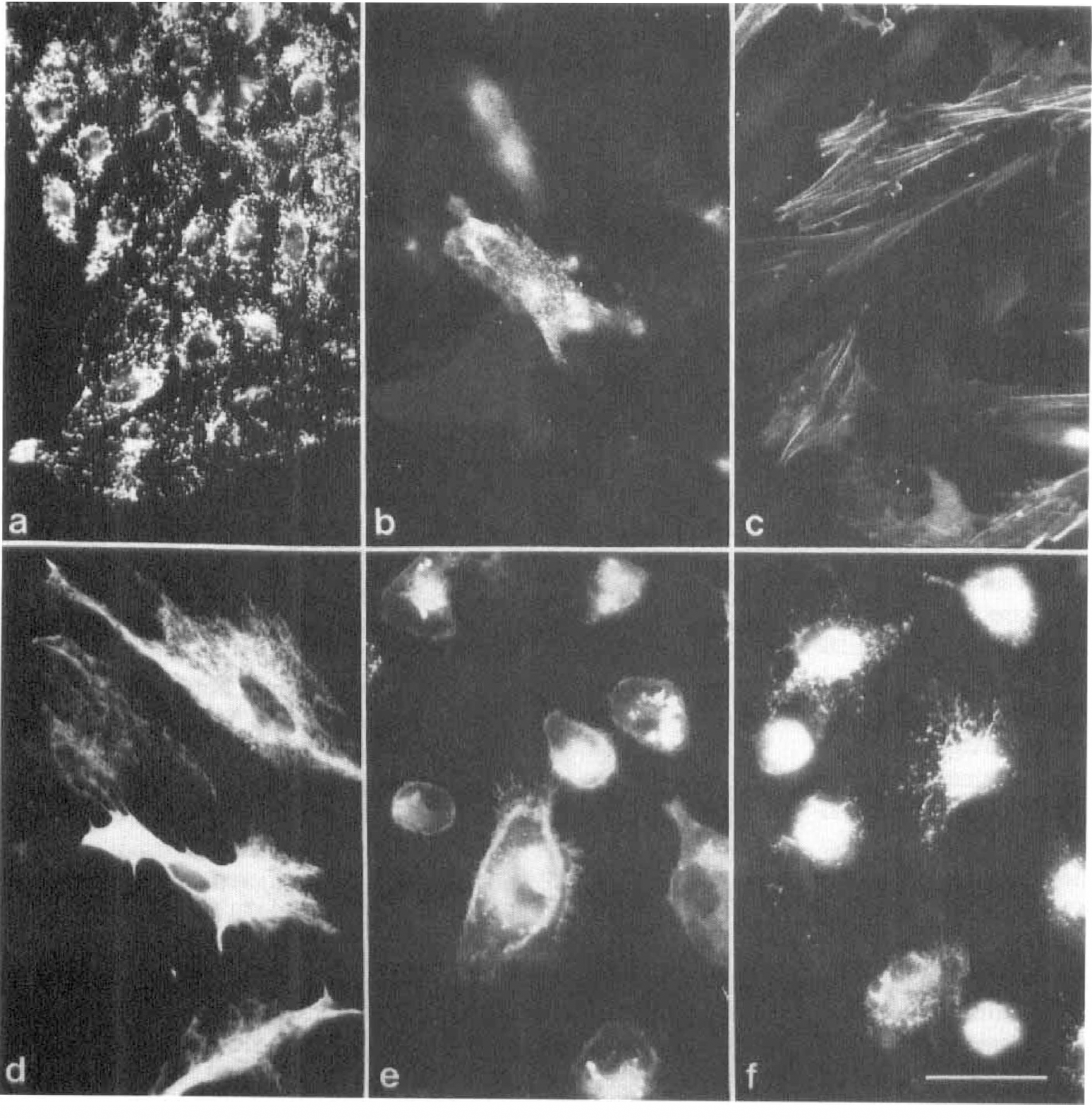
Immunofluorescent characterization of the crude microvascular cell fraction (CMF) after 20 days in vitro. A cluster of factor VIII-related antigen (F-VIII ra)-positive endothelial cells in primary culture are shown forming a typical contact-inhibited monolayer
Purified pericyte cultures. To obtain purified cultures of cerebral pericytes, we designed a solid-phase isolation technique using our mAb directed against the pericyte-specific enzyme pAPN. The isolation protocol consisted of immunoadsorption of antibody-labeled pericytes to immunomagnetic beads (for detail see Materials and Methods). The samples of immunoadsorbed cells contained a small number of individual pAPN-positive cells (Figs. 2a and 3a) and few microvascular segments (Figs. 2b and 3b). Contaminations with macrophages, free microglial cells, or astrocytes, as described above for the CMF, were not detected in PPC. Microvascular segments did not attach to the tissue culture supports and were lost after the first passage. After 15 to 40 days in culture, purified pericytes had grown in small islets (Fig. 2C) reaching area-restricted confluency (Fig. 2D) with the formation of nodules (Fig. 2E). The pericytic plaques resembled in both cellular morphology and their growth pattern the pericytes obtained from the CMF. All purified pericytes expressed sm-actin (Fig. 2F) and vimentin (Fig. 2G).
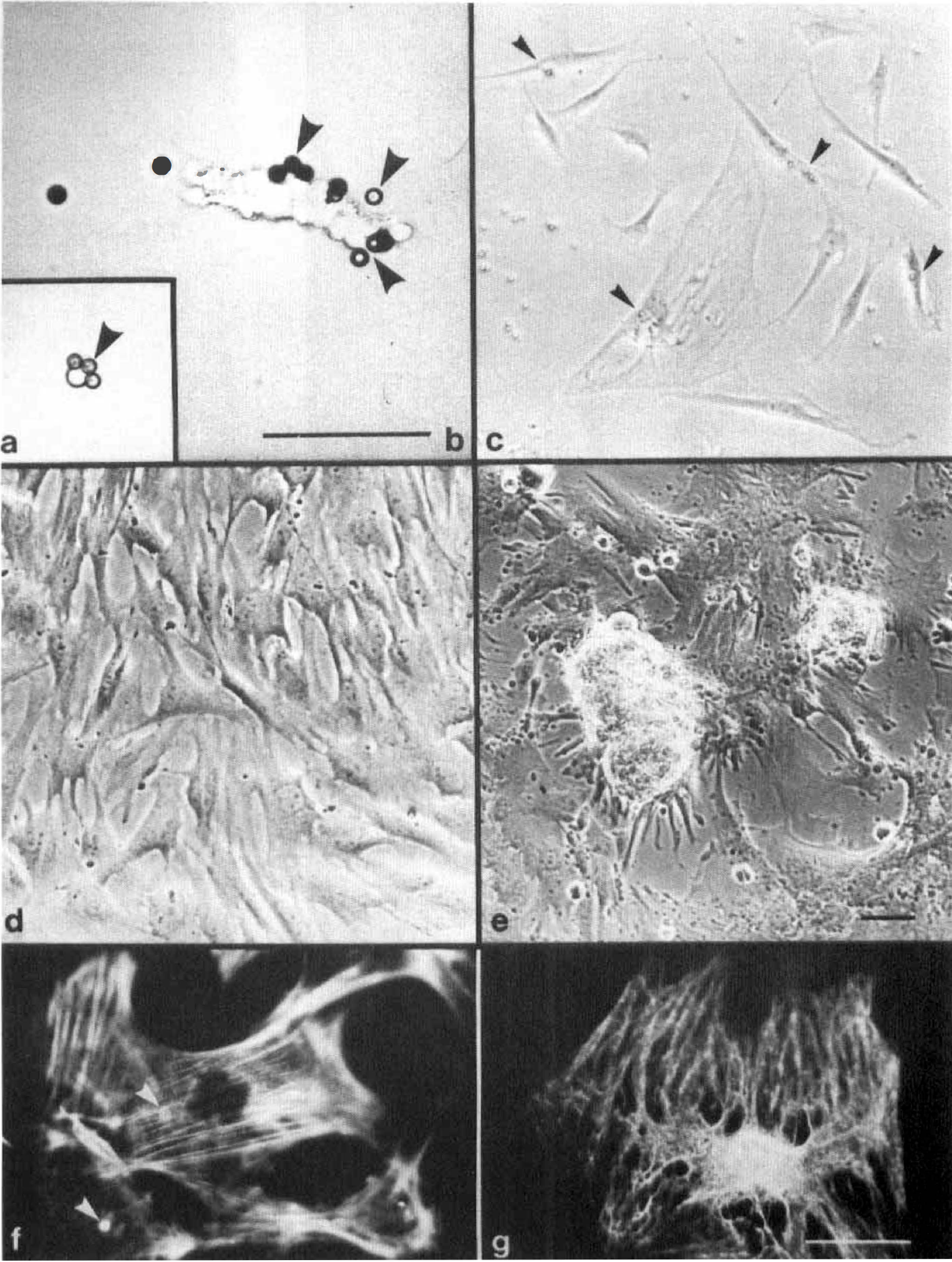
Isolation and culture of purified cerebral pericytes (PPC) achieved after separation by immunomagnetic beads. Acute isolated single cells
Immunofluorescent staining of PPC. Pericyte aminopeptidase N was expressed by acutely isolated pericytes
Mixed culture samples. The MCS consisted of the supernatants obtained after solid-phase adsorption of pericytes. In contrast to the PPC, the MCS revealed endothelial cells and contaminating macrophages, as well as microglial cells and very few astrocytes, but had in general a lower percentage of pericytes than the crude CMF (not shown).
pAPN protein expression is downregulated in vitro
The two cell fractions (PPC and MCS), which differed significantly in their cellular composition, were then exploited for studies of pAPN expression.
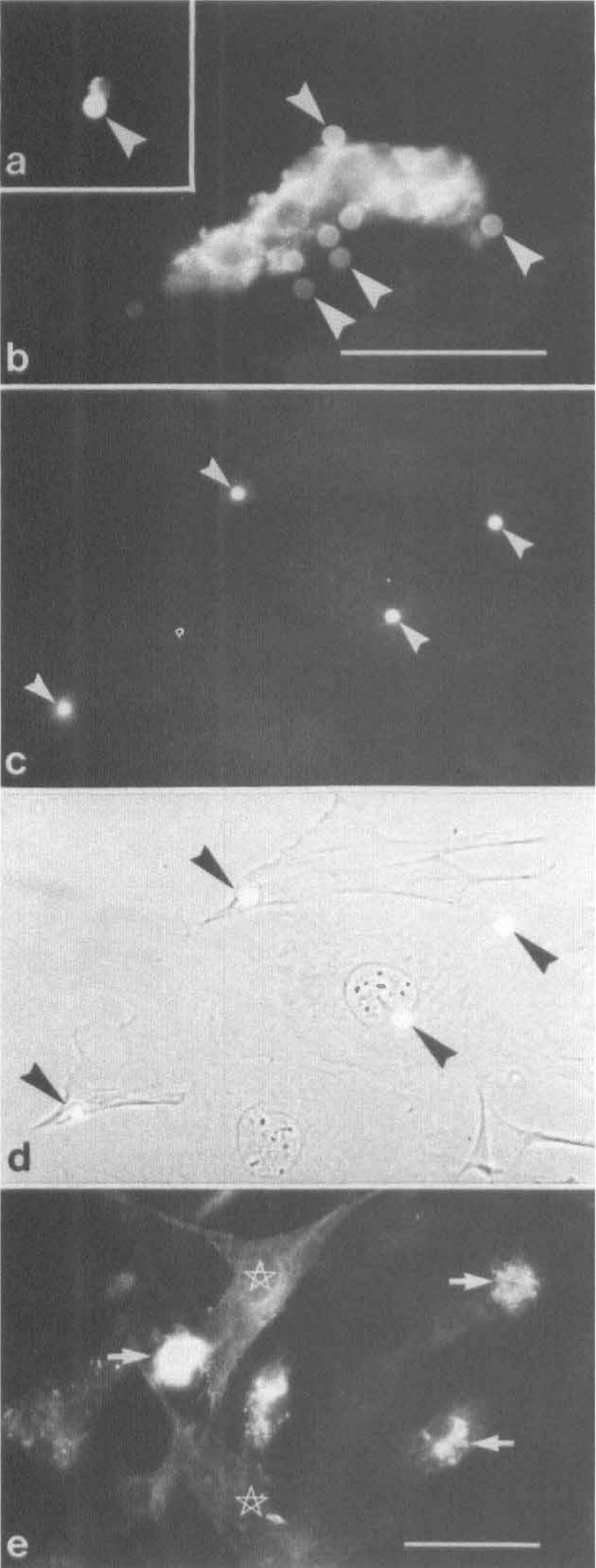
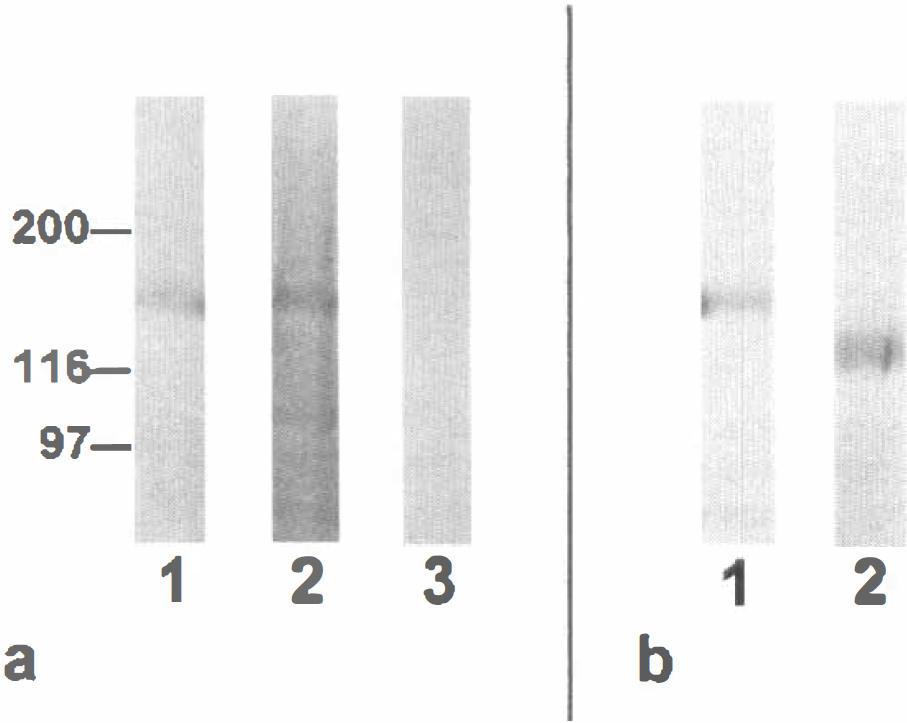
Expression of pAPN was high in acute isolated pericytes (Fig. 3A,B), as revealed by immunocytochemistry. In PPC, 2 to 5 days after plating, pAPN protein expression was completely downregulated as indicated by loss of pAPN immunoreactivity (Fig. 3C,D). In MCS, however, pAPN expression was still preserved at a lower level. Singular pAPN -positive cells were found in MCS even after long-term culture (>40 days; Fig. 3E). Expression and downregulation of pAPN appeared to be independent of the substrate used for both cell culture fractions (not shown). The downregulation effect was also confirmed by Western blots of freshly isolated microvessels and PPC (11 days in vitro). Purified pericyte cultures revealed no immunodetectable staining of pAPN as compared with the microvessel fraction (Fig. 4A).
To address the question of which level the downregulation of pAPN occurred on, we performed RT-PCR with subsequent Southern blotting of the pAPN amplicon (Fig. 5A) and in situ hybridization (Fig. 5B,C) to detect pAPN mRNA. Surprisingly both approaches revealed that mRNA of pAPN is still detectable in PPC and MCS even in long-term cultures (40 to 50 days in vitro, two passages).
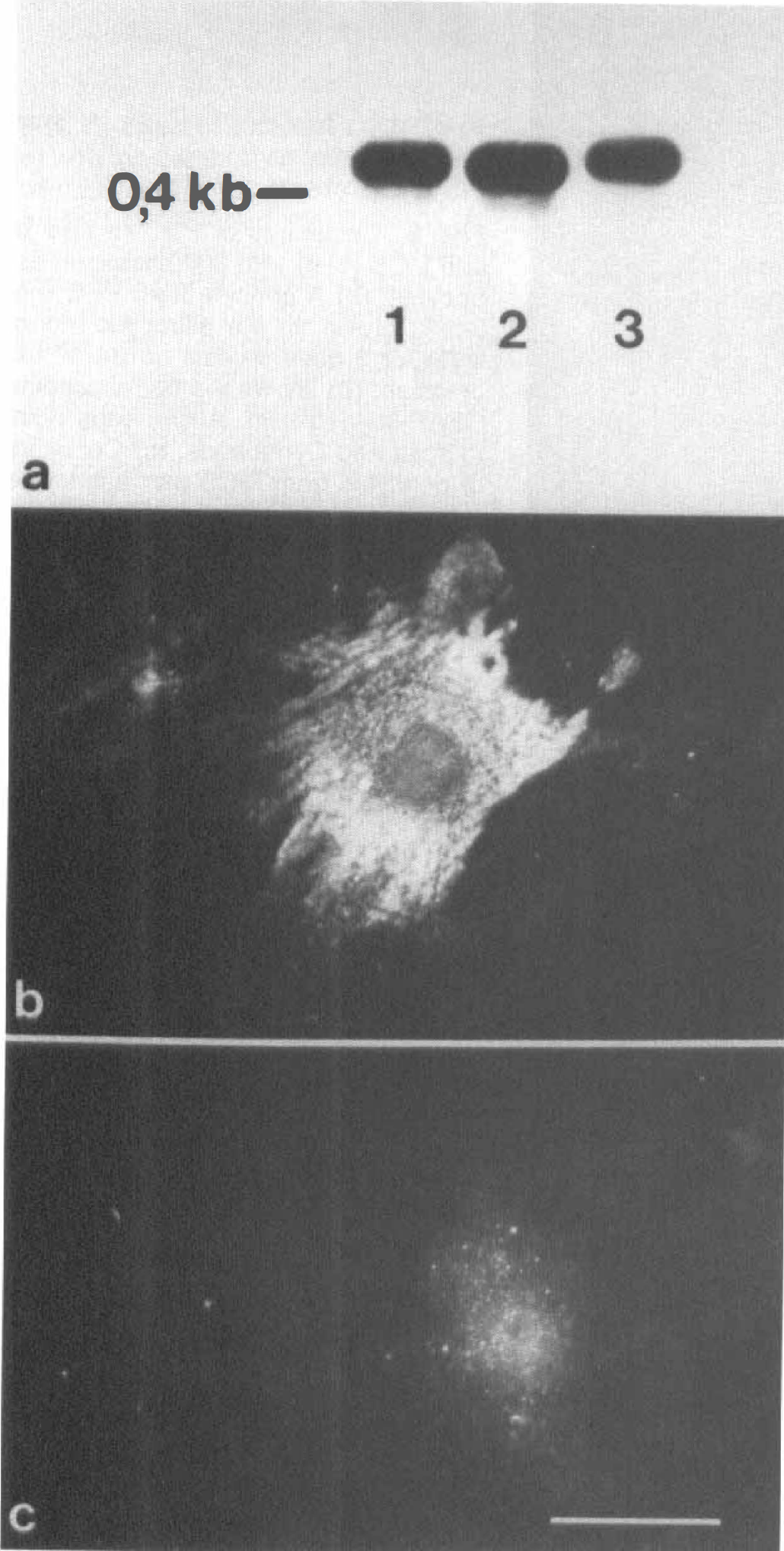
Detection of pAPN mRNA in long-term cultures of cerebral pericytes (>40 days). Southern blot analysis was performed using a digoxygenin-labeled reverse transcriptase polymerase chain reaction (RT-PCR) fragment amplified from RNA of acute isolated cerebrovascular cells
These findings strongly indicate that the regulation of pAPN expression is most likely to occur at the posttranscriptional level.
One reasonable possibility of posttranscriptional modification during tissue culturing is a change of the glycosylation pattern that is no longer detectable by the mAb. To exclude this possibility we performed endoglycosidase F digestion on kidney brush-border fractions and subjected the digested and undigested samples to Western blotting with the mAb. Kidney brush-border preparations were used to achieve a sufficient amount of digestible APN. As can be seen in Fig. 4B, glycosidase F treatment leads to an expected shift in APN migration but does not exert a loss of binding of the mAb.
Cocultures
Astrocytes have been documented to possess an inductive potency on the expression of BBB-specific features in cultured endothelial and pericytic cells (Maxwell et al., 1987, 1989; Cecchelli et al., 1992). We therefore investigated the effect of coculturing PPC and MCS with astrocytes on pAPN expression. When both preparations were kept in coculture with first-passaged astrocytes, no effect on pAPN expression in pericytes was achieved. In particular, PPC remained negative after 6 days of coculture (Fig. 6A,B), and MCS did not show any change in their low-level expression of pAPN (Fig. 6C—E). The lack of reinduction of pAPN expression in astrocytic cocultures was independent of the culture condition. Neither intimate contact in mixed cultures when both cell types were plated on one side of a glass support nor cultivation with limited cell-cell contact when cells were grown on separate sides of a polycarbonate filter led to a reappearance of pAPN immunodetectability (not shown). Apparently, pAPN expression in cerebral pericytes is not under the control of astrocytic factors.
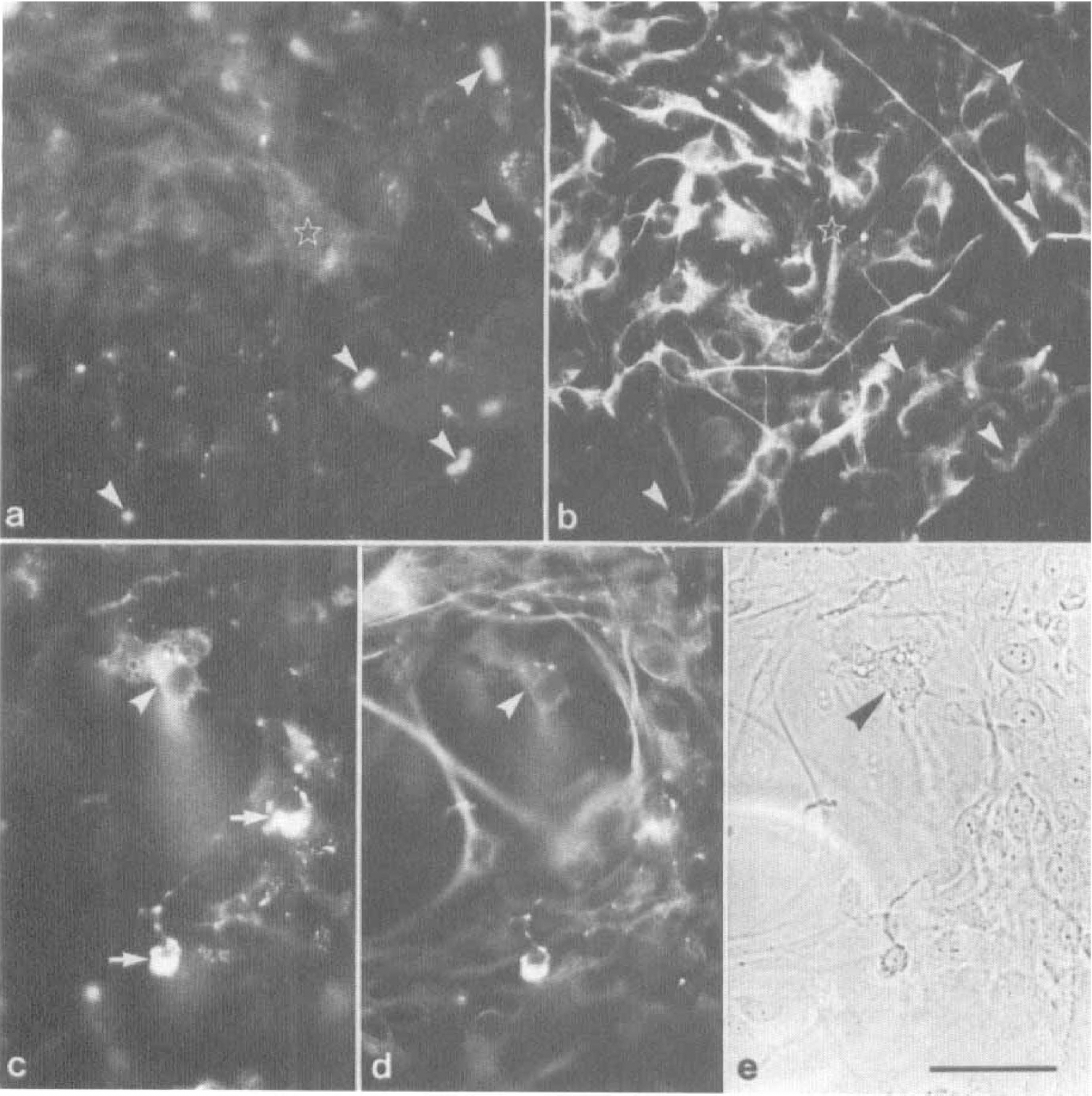
Cocultures of PPC
Alternatively, we used the brain endothelial cell line bEnd.3 for cocultures. In contrast to the astrocytic cocultures, bEnd.3 cells exhibited a significant effect on pAPN reexpression. After 40 days of culture, when MCS were transferred to socio-culture with bEnd.3 cells, a significant increase in the intensity of pAPN immunostaining was achieved (Fig. 7A,B). Frequency of pAPN-positive cells also increased by a factor of 10 as compared with the void MCS. To assess whether this effect was attributable to a humoral factor produced by the bEnd.3 cells, we cultured MCS in bEnd.3-conditioned medium. Conditioning of the medium was performed for 3 days. When MCS were cultured in the conditioned medium, the same effect was achieved as under coculture conditions (Fig. 7C,D).
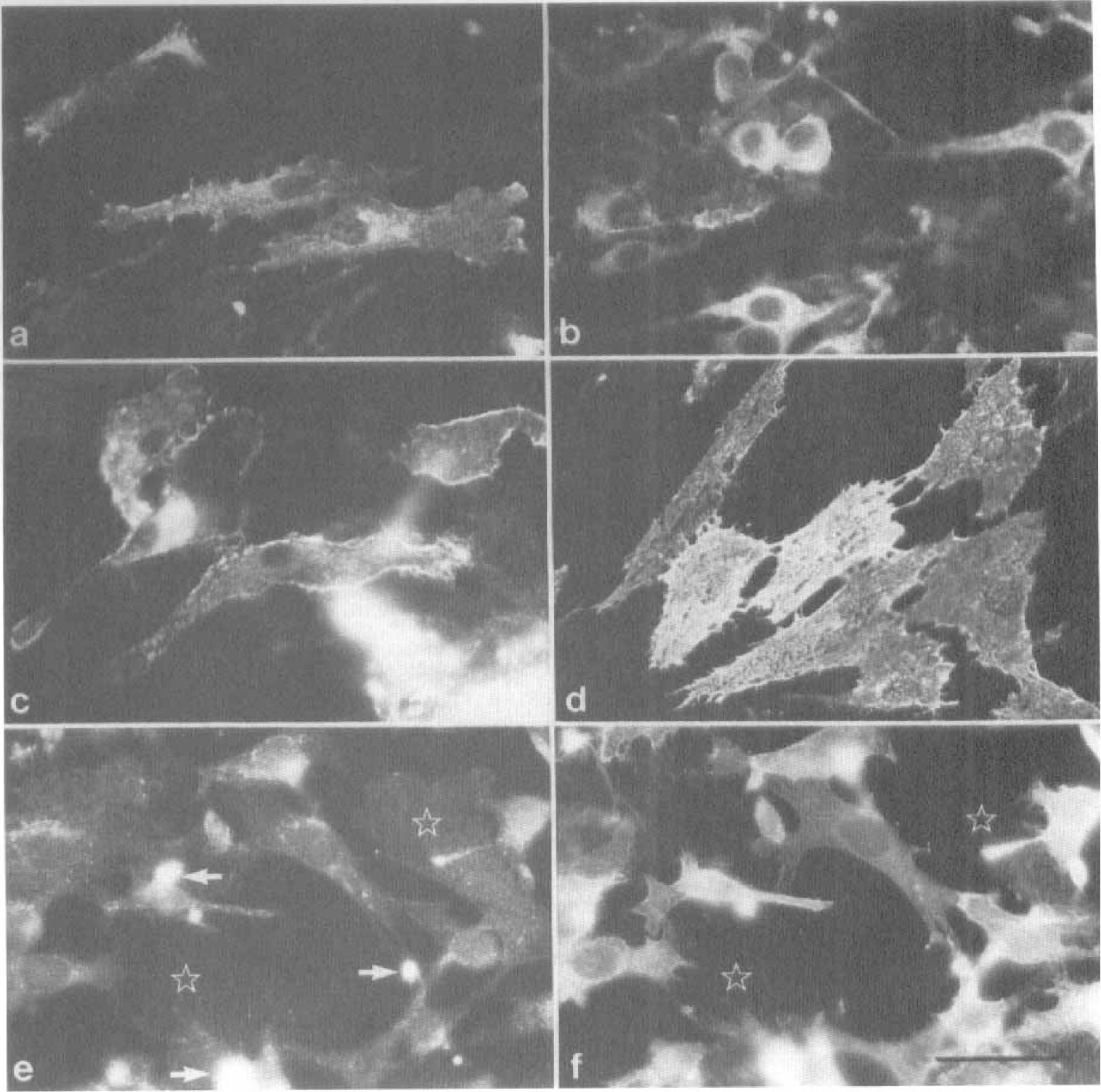
Effect of cocultures of MCS
Finally, we evaluated whether the in vitro history of the pericytes is essential for the humoral reinducibility of pAPN. For this purpose PPC were cocultured with bEnd.-3 cells. In cocultured pericytes deprived of endothelial cells no expression of pAPN was obtained (Fig. 7E,F). We therefore consider the permanent presence of coisolated cells, presumably endothelial cells, in the MCS responsible for the latent inducibility of pAPN expression in pericytes.
DISCUSSIONS
Evidence is accruing that the BBB is functionally and developmentally constituted by a multicellular complex involving endothelial cells as well as astrocytes, pericytes, and neurons, and that cooperative interaction among these cells is required to guarantee the functional integrity of the BBB (Dermietzel and Krause, 1991). This cooperation becomes most apparent when the maturation of the BBB during brain development is considered.
Morphogenetic factors required for the establishment of blood-brain barrier features
In the embryonal and postnatal brain, maturation of the BBB is considered to be guided by inductive and permissive interactions between pluripotent invading endothelial cells (Bär, 1980) and its neuroectodermal environment (Risau, 1995; Stewart and Wiley, 1981). According to this paradigm, several studies revealed a differentiation of BBB features and the onset of brain-specific endothelial protein expression concurrent with the differentiation of the brain (Qin and Sato, 1995; Dermietzel et al., 1992; Risau et al., 1986a, 1986b). Apparently, the final acquisition of the BBB depends on a morphogenetic program that involves the cooperation of several cellular components. Information on the molecular factors participating in these processes are rare but a recent example dealing with the expression of the vascular endothelial growth factor-2 (VEGFR-2) receptor provides support for the idea that a temporal morphogenetic regimen governs BBB maturation. Although the expression of VEGFR-2 is high during brain angiogenesis, which necessitates the potent mitogenic activity of VEGF, the receptor density is downregulated when BBB maturation is initialized (Plate et al., 1994), a necessary prerequisite for the acquisition of BBB tightness because VEGF is known to be a vascular permeability factor (Millauer et al., 1993; Ferrara et al., 1992).
With respect to the cellular entities primarily involved in BBB induction, astrocytes have been considered to represent the key cellular component, both in situ (Janzer and Raff, 1987) and in vitro (see introduction). However, recent investigations have challenged the simplistic view of astrocytic induction of the BBB. Immature neuroepithelial cells or glial progenitor cells are now considered to be responsible for the initiation of BBB development (Holash et al., 1993).
In this context, the role of pericytes in BBB differentiation and establishment should be reconsidered. Recent in vitro studies have shown that pericytes and endothelial cells of CNS origin have mutual influence on each other's replicative and biosynthetic behavior. Retinal capillary endothelial cells, for example, secrete a heparin-binding factor that promotes pericyte proliferation (Swinscoe and Carlson, 1992). On the other hand, coculturing endothelial cells with pericytes results in the activation of a latent form of transforming growth factor-β1 (TGF-β1) and in the subsequent suppression of endothelial cell growth (Orlidge and D' Amore, 1987). However, TGF-β1 also increases endothelin-1 expression in vascular endothelium, which in turn operates as a pericytic mitogen and causes contraction of pericytes (Chakravarthy et al., 1992). These results suggest complex biologic feedback mechanisms between endothelial cells and pericytes.
Cerebral pericytes, like endothelial cells, are substituted with a specific set of proteins that are functionally involved in the enzymatic component of the BBB. Aminopeptidase A (Healy and Wilk, 1993; Song et al., 1993), γ-glutamyl transpeptidase (Risau et al., 1992), and pAPN (Kunz et al., 1994) are well-studied representatives of the pericytic enzymatic barrier, which is primarily involved in proteolytic activities.
Pericyte aminopeptidase N has recently been revealed to be a late BBB marker in rodent brain development occurring around embryo day 18 of brain angiogenesis (Dermietzel and Krause, 1991). Thus, the final establishment of the pericytic BBB feature can be considered to succeed in most of the endothelial BBB components, in particular, in the achievement of endothelial tightness (Dermietzel and Krause, 1991; Bauer et al., 1992).
In vitro expression of pAPN requires a soluble endothelial factor and depends on coculturing conditions
To gain more information on the inductive mechanisms involved in the expression of this prominent BBB enzyme, we studied the expression of pAPN under various culture conditions.
Downregulation of pAPN occurred within 2 to 3 days when pericytes became adherent. However, as shown by RT-PCR and subsequent Southern blotting, mRNA for pAPN remained detectable even in long-term cultures. This finding strongly suggests a posttranscriptional regulation for pAPN expression in vitro. One reasonable explanation for the loss of detectability of pAPN is a stress-induced switch in the glycosylation pattern of pAPN (O'Connell et al., 1990), resulting in a modified isoform that is no longer accessible to our mAb.
However, this possibility could be ruled out by glycosidase treatment of an APN-enriched membrane fraction. Kidney cells of the proximal tubules that have been shown to express the same isoform of APN (Kunz et al., 1994; Watt and Yip, 1989) showed no loss of binding of the mAb after glycosidase digestion when subjected to Western blotting. These findings are indicative for a persistent recognition of the pAPN epitope by the mAb. It seems therefore more likely that APN gene expression is controlled by posttranscriptional regulation mechanisms, as has been shown recently for the same enzyme in T lymphocytes, natural killer cells, and permanently growing tumor cells (Wex et al., 1997).
By using coculture systems of purified pericytes with both primary astrocytes and immortalized endothelial cells, we further examined the possible inductive and modulating effects of cellular components on pAPN expression. The most important finding of these experiments revealed that pAPN expression is enhanced by one or more soluble endothelial factors. Interestingly, this inductive effect depended on the socio-culture conditions of the pericytes. Only when mixed pericytic cultures were grown in coculture with endothelial cells could pAPN expression be significantly enhanced by endothelial cell-conditioned medium. Purified pericytes grown in complete isolation from endothelial cells revealed no pAPN reexpression potency at all. Although primary astrocytes from neonatal rat brains have been described to induce various endothelial BBB features in vitro (see above), we found no effect on pAPN expression in our cell culture model. A reasonable explanation for this effect is that the differentiation program of pericytes is under control of endothelial cells and, moreover, that a deprivation of pericytes from this supportive “string” leads to an irreversible loss of pericytic differentiation potency. Thus, our experimental design provides stringent in vitro evidence that BBB differentiation requires the cooperation of different vascular cellular components and that the achievement of functionally differentiated cerebral pericytes is closely related to endothelial cells.