
Abstract
Tissue adenine nucleotides are depleted during cerebral ischemia, impeding recovery after reperfusion. Although prior studies have attempted to prevent the initial loss of adenylates, the present study tests the hypothesis that stimulating synthesis of adenine nucleotides, through either adenosine kinase or adenine phosphoribosyltransferase, would result in significant cerebroprotection. To study the effects on neurons and glia directly while avoiding the influence of the cerebral vasculature, hippocampal brain slices were used for the model of transient ischemia with reperfusion. The standard brain slice insult of brief exposure to anoxia with aglycemia was modified based on studies which showed that a 30-minute exposure to air with 1 mmol/L glucose produced a stable, moderate reduction in ATP during the insult and that, 2 hours after return to normal conditions, there was moderate depletion of tissue adenine nucleotides and histologic injury. Treatments with 1 mmol/L adenosine, AMP, or adenine were equivalent in partially re-storing adenine nucleotides. Despite this, only adenosine af-forded histologic protection, suggesting a protective role for adenosine receptors. There also was evidence for metabolic cycling among adenine nucleotides, nucleosides, and purines. Adenosine may exert direct cerebroprotective effects on neural tissue as well as indirect effects through the cerebral vasculature.
The degradation of ATP to AMP and on to adenosine during cerebral ischemia presents a dilemma for acute stroke management. On the one hand, this has the negative effect of reducing the amount of total adenine nucleotides (TAN) so that, after reperfusion, even if the adenylate energy charge (AEC) returns promptly to normal, TAN frequently remains low, limiting the potential flux of high-energy phosphate bonds necessary for recovery (Bereczki and Csiba, 1993; Folbergrova et al., 1995; Sun et al., 1995). On the other hand, as AMP increases, adenosine is released into the extracellular space (ECS) (Van Wylen et al., 1986; Morimoto et al., 1991; Matsumoto et al., 1993; Phillis et al., 1994) where it appears to play an important role in regulating CBF and as a cerebroprotective agent during and after ischemia (Fredholm, 1997). Balancing these two competing consequences of ATP degradation poses a significant challenge.
Strategies for limiting the TAN depletion can follow one of three broad categories, either slowing the enzymatic conversion of adenine nucleotides to nucleosides pharmacologically, inhibiting adenosine transport, or replenishing adenine nucleotides by providing nucleoside or purine precursors (Fig. 1). The danger of the first approach is that limiting production of adenosine may reduce its cerebroprotective effect. Limiting loss of adenine nucleotides is attractive as a strategy because this would avoid the need for re-synthesis and would reduce the expenditure of high-energy phosphate bonds just when they are least available. The approach is worth pursuing because it is not yet clear how much adenosine must be released for cerebroprotection to occur. Also, conversion of even a minor percentage of tissue ATP can produce a large relative increase in adenosine because, under normal conditions, the concentration of ATP is about 50 times higher than that of adenosine (Fredholm, 1997). Several approaches to avoiding enzymatic depletion of AMP have been tested in a variety of stroke models. Blocking the conversion of adenosine to inosine via the enzyme adenosine deaminase consistently increases adenosine outflow during ischemia while reducing the outflow of inosine and hypoxanthine (Phillis et al., 1991). Although this strategy does increase tissue ATP (Phillis and O'Regan, 1996), attempts at cerebroprotection have been disappointing thus far (Lin and Phillis, 1992; Turkozkan et al., 1996). The effects of blocking adenosine kinase have been similarly disappointing (Phillis and Smith-Barbour, 1993). While blockade of 5'-nucleotidase can increase adenosine release during ischemia (Pedata et al., 1993), no studies have directly addressed cerebral neuroprotection by this approach. More promising results have been obtained by blocking bidirectional adenosine transport with propentofylline or dipyridamole. This approach, which increases extracellular adenosine while reducing conversion of adenosine to inosine and hypoxanthine (Fredholm et al., 1994; Park and Gidday, 1990; Fowler, 1993), has been found to reduce infarct volume in at least two animal models (Park and Rudolphi, 1994; Kano et al., 1994). The mechanism remains uncertain, however, because propentofylline does not increase adenine nucleotide levels during reperfusion (Phyllis and O'Regan, 1996) and does interact with both A1 and A2 receptors (Parkinson and Fredholm, 1991).
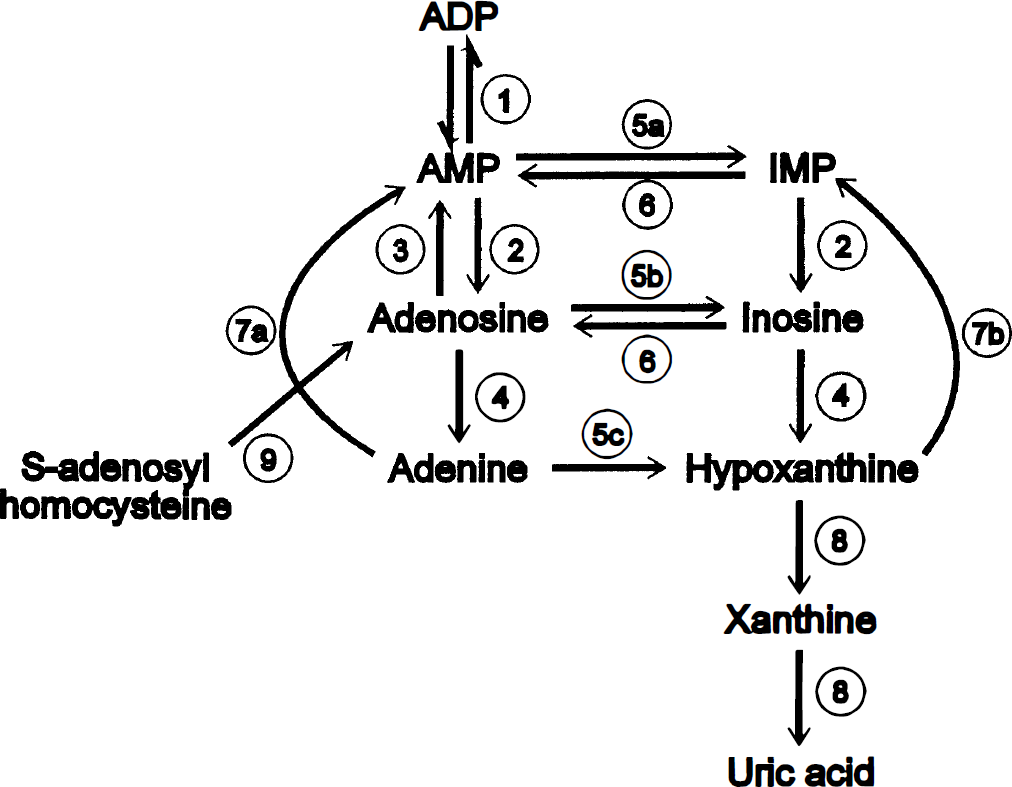
Metabolic pathways of adenine nucleotide degradation and synthesis. Enzymatic reactions are represented by circled numbers, reversible reactions as paired single-headed arrows and unindirectional reactions by double headed arrows. 1, nucleoside monophosphate kinase; 2, 5'-nucleotidase; 3, adenosine kinase; 4, nucleoside phosphorylase; 5a, adenylate deaminase; 5b, adenosine deaminase; 5c, adenine deaminase; 6, sequential reactions of adenylsuccinate synthetase followed by adenylosuccinase; 7a, adenine phosphoribosyltransferase; 7b, hypoxanthine phosphoribosyltransferase; 8, xanthine, oxidase; 9, S—adenosylhomocysteine hydrolase.
Less attention has been given to the strategy of stimulating re-synthesis of adenine nucleotide compounds after ischemia. It has been shown in brain slices that supplementing the buffer with adenosine does increase total tissue adenine nucleotides and ATP content (Whittingham et al., 1989) and that 3H-adenine can be used to label ATP (Fredholm et al., 1994). It has also been observed that idebenone enhances formation of adenine nucleotides in brain slices during “reperfusion” of slices exposed to anoxia, perhaps by stimulating conversion from S-adenosylhomocysteine (Latini et al., 1993). Recently, strong evidence has emerged indicating active cycling of adenine compounds during and after ischemia through pathways involving both adenosine kinase and the purine salvage pathway in red blood cells (Smolenski et al., 1991), heart (Zoref-Shani et al., 1992), and liver (Bontemps et al., 1993), as well as in brain (Morimoto et al., 1991). However, no direct studies have shown a cerebroprotective effect of exogenous adenosine or other compounds that stimulate the synthesis of adenine nucleotides. There is also the potential interpretive problem of separating the beneficial role of adenosine as a neuromodulator from any direct effect of adenosine as adenine nucleotide precursor.
Our goal is to study cerebroprotection related to the re-synthesis of adenine nucleotides in an in vitro model of cerebral ischemia and reperfusion. The ideal model should realistically reflect in vivo cerebral ischemia in terms of the duration and severity of metabolic insult, be reproducible, and be capable of identifying strategies with potential for cerebroprotection in humans during stroke. Specifically, the model should consistently produce a decrease in AEC to between 0.4 and 0.5 during the insult and show a 50% reduction in TAN with moderate histologic injury after 2 hours of reperfusion. A model meeting these conditions would be at once similar to in vivo ischemia (Komatsumoto et al., 1987; Folbergrova et al., 1992; Wagner et al., 1992) and experimentally practical. The standard brain slice model of “ischemia and reperfusion” exposes brain slices to anoxia with aglycemia for 5 or 6 minutes. However, this model has the disadvantages that it produces a very severe depletion of ATP (Whittingham et al., 1984), tends to produce an all or none injury (Taylor et al., 1985; Schurr et al., 1986; Weber and Taylor, 1994), and does not show protection even after adenosine supplementation is used to increase tissue adenine nucleotide and ATP content (Whittingham et al., 1989). In the present study, we have systematically studied the effects of varying degrees of O2 and glucose deprivation on hippocampal brain slices as well as insult duration using histology and HPLC measurements of compounds related to adenine nucleotides. We then used the optimal model to assess the metabolic and histologic effects of 1 mM adenosine, AMP, and adenine.
METHODS
Materials
Inorganic salts, adenine HC1, and the Na salts of adenosine and AMP were of cell culture grade (Sigma Chemical Co., St. Louis, MO) while dextran was industrial grade. All gases were purchased from Liquid Carbonics (Bethlehem, PA). Nucleotide and nucleoside standards were purchased from Sigma and KH2PO4, methanol and acetonitrile, all HPLC grade, were purchased from Fisher (Springfield, NJ).
Brain slice incubations
Brain slice procedures were the same as those described previously (Newman et al., 1995). Briefly, male 250-g Sprague-Dawley rats were anesthetized (3% halothane—70% NO2/30% O2) and then cooled on ice to 30°C (Newman et al., 1992; Español et al., 1994). After decapitation, the brain was rapidly removed and chilled briefly. Both hippocampi were removed and chopped at 450 μm on a Smith-Farquhar tissue chopper. Twelve slices were collected and placed in incubation chambers designed for biochemistry and histology (Newman et al., 1995) at 22°C for 45 minutes and then gradually warmed to 37°C over 30 minutes and maintained at that temperature for the remainder of the experiment. Control incubation buffer was (in mmol/L): NaCl, 122; MgSO4, 1.3; NaHCO3, 21; KH2PO4, 1.2; KC1, 3.0; glucose, 5; and CaCl2, 1.5; gassed with 95% O2—5% CO2, at pH 7.38±0.02. Buffer also contained 3.1% dextran (MW ∼77,000), flowed at 1 mL/min, and was re-equilibrated with gas flowing at approximately 90 mL/min on entry into the incubation chamber. All slices were incubated in this control buffer for the first 75 minutes in vitro after which the buffer was changed to the experimental buffer. Experimental buffer changes differing from control by more than 1 mOsm were osmotically compensated by alteration of NaCl.
“Ischemic” insults were initiated by transferring rings, with either three slices for histology or six slices for adenine nucleotides, into chambers pre-equilibrated with the appropriate buffer and gas. Reperfusion was initiated by transferring rings from insult chambers into separate chambers equilibrated with control buffer at 95% O2 for studies related to development of the reperfusion model. For adenine nucleotide recovery experiments, slices were exposed to insult and then returned to control buffers equilibrated with 95% O2 or to control buffers containing 1 mmol/L AMP, 1 mmol/L adenosine or 1 mmol/L adenine. O2 tension was modified by bubbling buffer with 95% N2—5% CO2 for 0% oxygen, air—5% CO2 for 20% oxygen, and by bubbling with a mixture of 95% O2—5% CO2 and air—5% CO2 for 40% and 50% oxygen. These mixtures were achieved by combining the two gas streams, air and oxygen, at carefully regulated outlet pressures of 15 psi and altering the flow rate of each stream using Gilmont adjustable flowmeters to obtain the desired O2 tension. Control slices for adenine nucleotide recovery experiments were lifted in their rings and returned to 95% O2—5% CO2 with 5 mmol/L glucose for 30 minutes and then lifted again to continue in control conditions for an additional 2 hours so that all slices were exposed to the same degree of manipulation. For adenine nucleotide recovery studies, 1 mmol/L adenine, adenosine and AMP were added as powders to control buffer, with osmotic compensation.
Analytical methods
Slices for histology were incubated for a total of 2 hours of reperfusion, immersion-fixed in Bouin's fixative at 37°C for 1 hour, stored in 70% ethanol, dehydrated, paraffin embedded, sectioned at 7 μm and stained with hematoxylin and eosin. All sections were collected and inspected visually, and the center section was scored using a subjective, ordinal neuronal scale (1—all neurons normal; 2—most normal; 3—evenly mixed normal and abnormal; 4—most neurons injured, some destroyed; 5—no normal tissue, most neurons destroyed). Five regions were analyzed: CA1, CA2, CA3, CA4, and the inner blade of dentate.
Slices for adenine nucleotides were fixed by freezing them to the walls of microfuge tubes chilled in liquid N2. The tissue was sonicated, using a Microson XL Ultrasonic Cell Disruptor with a microprobe, into 150 μL of 0.3 mol/L perchloric acid, centrifuged at 12,000 × g at 4°C for 20 minutes to form a protein pellet. The supernatant was then neutralized with 15 μL of 3 mol/L KHCO3 and analyzed immediately. ATP, ADP, AMP, adenosine, inosine, adenine, and hypoxanthine were resolved by a modification of the HPLC method of Teerlink et al. (1993). The separations were performed on a Bio-Rad 2800 HPLC system with an autosampler and a ultraviolet spectrometer set to 210 nm. A 50-μL aliquot of the neutralized acid extract was injected onto a MicroSpher C18 reverse phase column (Chrompack, Raritan, NJ) and eluted at 1 mL/min with 0.15 mol/L KH2PO4 at pH 5.0 as solution A and water:acetonitrile:methanol (50:25:25) as solution B. The gradient was as follows: 0% B for 1 minute, linear ramp to 15% B at 5.4 minutes, linear ramp to 45% B at 6.4 minutes, hold at 45% B until 7.4 minutes, linear ramp down to 0% B at 8.0 minutes and re-equilibrate at 0% B until 12 minutes. All peaks were quantified by peak height with reference to standard curves run each day at the start and end of the sample set. All metabolites were normalized for slice protein (Lowry et al., 1951). Measurements of AMP for slices incubated in AMP, of adenosine for slices incubated in adenosine, and of adenine for slices incubated in adenine were corrected by subtracting the extracellular contributions using our previously measured ECS of 0.254 mL/g (unpublished data) and assuming that the concentration in the ECS was the same as that in buffer. We also calculated
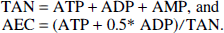
Statistical analyses were performed using SPSS/PC+ for DOS, version 5.0.2 (SPSS, Inc). Because of the multiple comparisons involved in all experiments, minimal significance was chosen as P < .01. ATP, ADP, AMP, and AEC were analyzed by multivariate analysis of variance (MANOVA) with multiple range tests by the least significant difference method and subsequent univariate analyses when MANOVA revealed statistical significance. TAN is a linear combination of ATP, ADP, and AMP and cannot be analyzed by MANOVA. Histology was analyzed both as parametric data with analysis of variance (ANOVA) and t-tests and as nonparametric data with Kruskal-Wallis one way ANOVA and Wald-Wolfowitz runs tests. Values are reported as means±SD.
RESULTS
Development of reperfusion model
Anoxia induces a severe metabolic insult in slices. During exposure to anoxia with 5 mmol/L glucose (Fig. 2A), slice ATP decreases from 8 to less than 4 nmol/mg protein within 3 minutes. AMP increases from less than 1 to more than 5 nmol/mg protein by 6 minutes, with large increases already apparent within 3 minutes. Levels of ADP remain stable. AEC decreases from 0.8 to 0.4 by 4 minutes and to 0.3 within 7 minutes (Fig. 3). TAN decreases only slightly from the control value of 12.0±1.1 during this acute insult. The insult is even greater when slices are exposed to anoxia with 1 mmol/L glucose (Fig. 2B). Slice ATP decreases to less than 2 nmol/mg protein and AMP reaches 5 nmol/mg protein within 3 minutes, with a small decrease in ADP as well. AEC is less than 0.3 within 4 minutes and stabilizes at about 0.2 (Fig. 3). TAN decreases by 25% within 7 minutes of starting the insult. Although there is no apparent histologic injury during the acute phase of anoxia (Fig. 4A), when slices are exposed to anoxia and then returned to normal oxygenation for 2 hours, there is a relatively abrupt transition toward severe injury as insult duration increases from 4 to 6 minutes (Fig. 4B). Minor histologic injury of CA1 and dentate is apparent with insults as brief as 4 minutes, but all regions of the slice show injury as the insult increases to 6 minutes. Thus, it appears that anoxia induces a severe metabolic insult with significant tissue injury after just 6 minutes even at 5 mmol/L glucose. Decreasing glucose to 1 mmol/L worsens the metabolic disturbances substantially, compressing the time course still further.
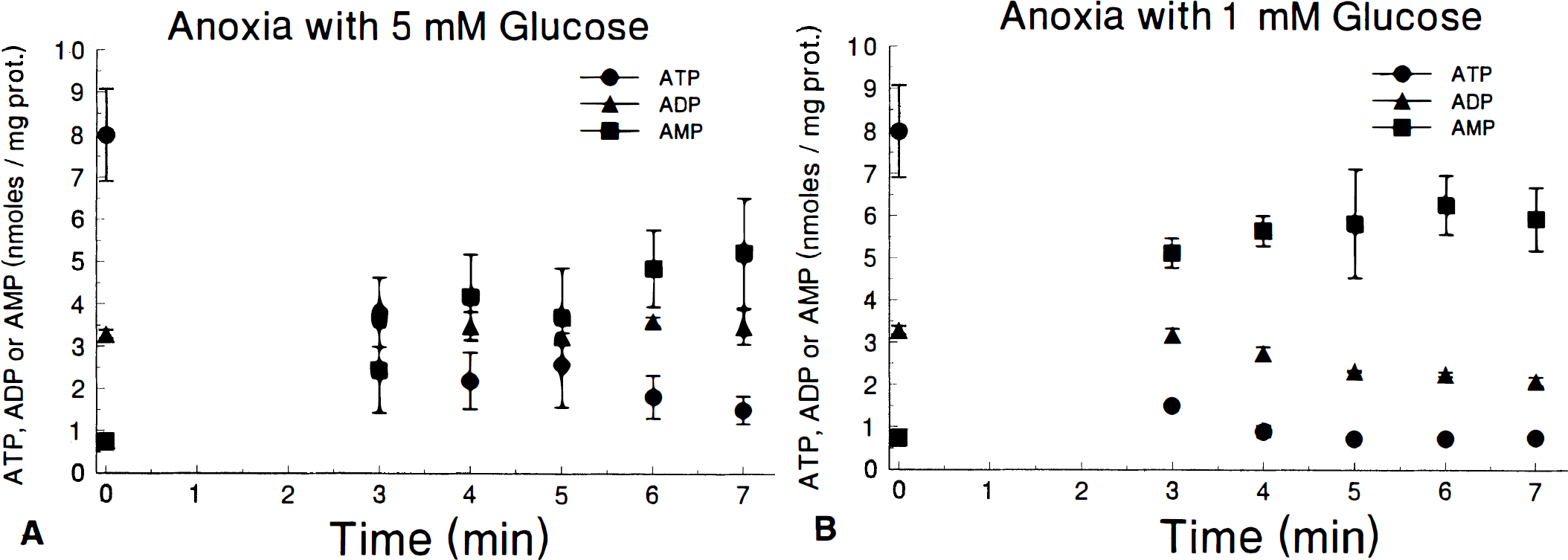
Time course of slice adenine nucleotides during exposure to anoxia with 5 mmol/L (
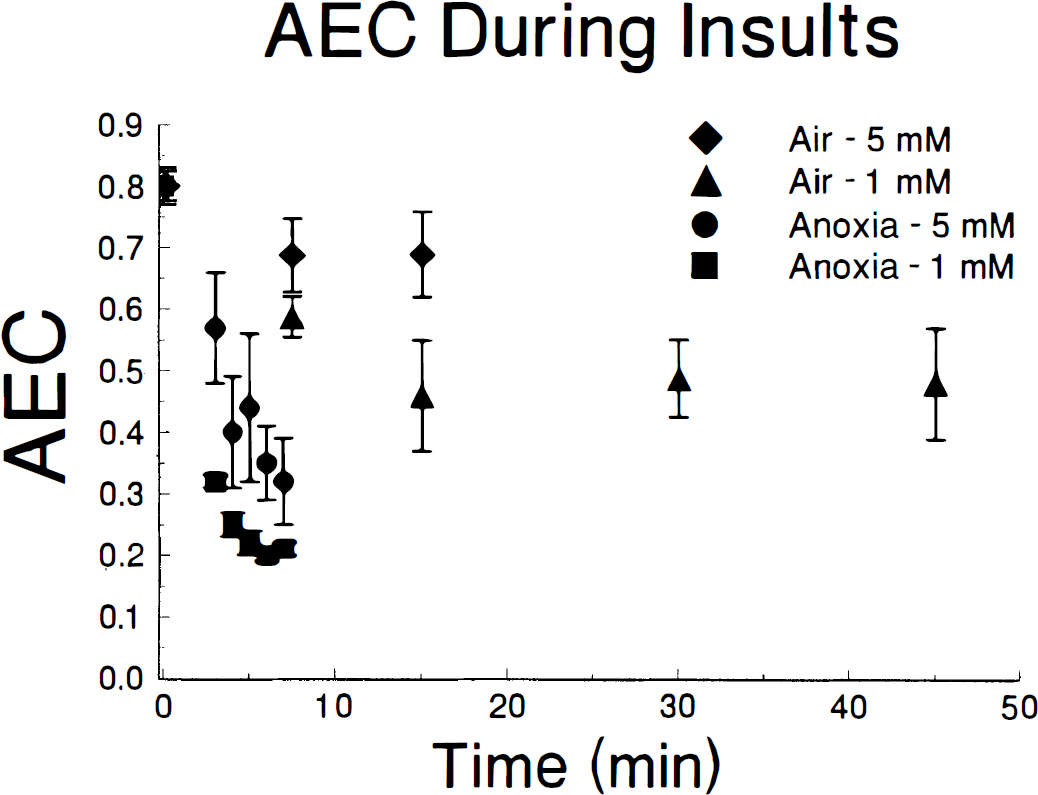
Time course of slice adenylate energy charge during continuous exposure to buffer equilibrated with 95% N2—5% CO2 (anoxia) or 95% air—5% CO2 with either 5 mmol/L or 1 mmol/L glucose.
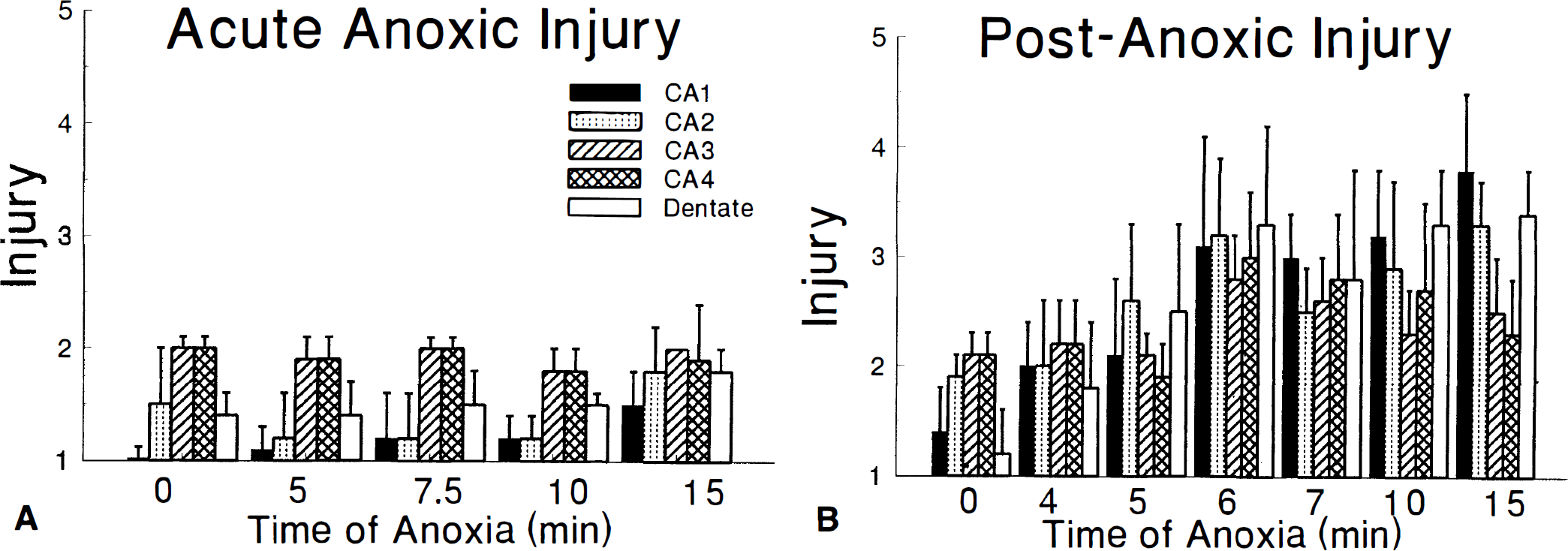
Histologic injury resulting from exposure to buffer equilibrated with 95% N2—5% CO2. (
Slices exposed to 50% O2 with 5 mmol/L glucose show little change in any of the adenine nucleotides despite prolonged insults (18 slices studied, data not shown). AEC decreases to 0.7 after 30 minutes and then is stable for up to 150 minutes. TAN is unchanged from controls after 30 minutes at 50% O2 and decreases by less than 25% by 150 minutes of continuous exposure to this degree of hypoxia. Decreasing oxygen to 40% with 5 mmol/L glucose (17 slices studied, data not shown) causes AEC to decrease to 0.6 at 30 minutes and then decrease further, to 0.5 by 90 minutes and to 0.4 after 150 minutes of continuous insult. During the same period, TAN decreases gradually by a total of 40% after 150 minutes. Therefore, both insults appear too mild for producing a reperfusion model. Insults with 40% O2 have the further disadvantage of being unstable throughout the exposure time. These results, and the results with anoxia, suggested that exposing slices to 20% O2 as air might be suitable as a basis for studying reperfusion injury.
The metabolic insult incurred when brain slices are exposed to air—50% CO2 is highly dependent on the buffer glucose concentration (P < .001; Figs. 3 and 5A). There are only insignificant changes in adenine nucleotides when slices are exposed to air with 5 mmol/L glucose but, as glucose is reduced from 3.5 to 1 mmol/L, there are very large decreases in ATP and increases in AMP with significant glucose dependence for both AEC and TAN as a consequence. At 0.5 or 0 mmol/L glucose, AMP reaches 5 nmol/mg protein while ATP decreases to less than 1.5 nmol/mg protein, so that AEC reaches 0.32±.03 after 15 minutes. TAN decreases slightly over the first 15 minutes of exposure to air with 0 mmol/L glucose to reach 9.8±.9. Slices exposed to air with 1 mmol/L glucose rapidly achieve an AEC between 0.4 nd 0.5 (Fig. 3). In addition, the concentration of adenosine accumulating in slices varies with glucose concentration (Fig. 6A), reaching the highest levels at 0.5 and 0 mmol/L glucose. There are no significant effects of glucose on inosine, with a mean of 0.10±0.01 nmol/mg protein over all glucose concentration, or of hypoxanthine, with a mean of 0.22±.25 nmol/mg protein. There are, however, significant effects of glucose on tissue adenine (P < .008) although this dependence is eliminated (P = .259) when the dependence is analyzed using tissue adenosine as a covariate.
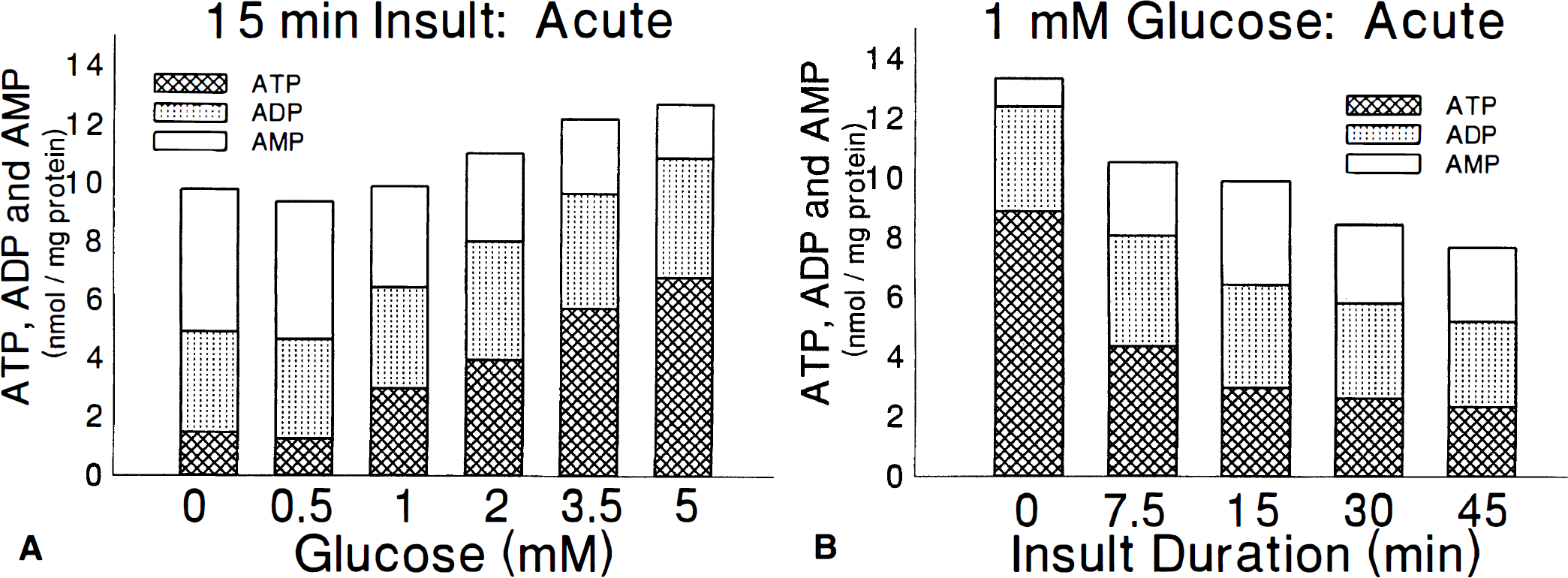
Influence of glucose
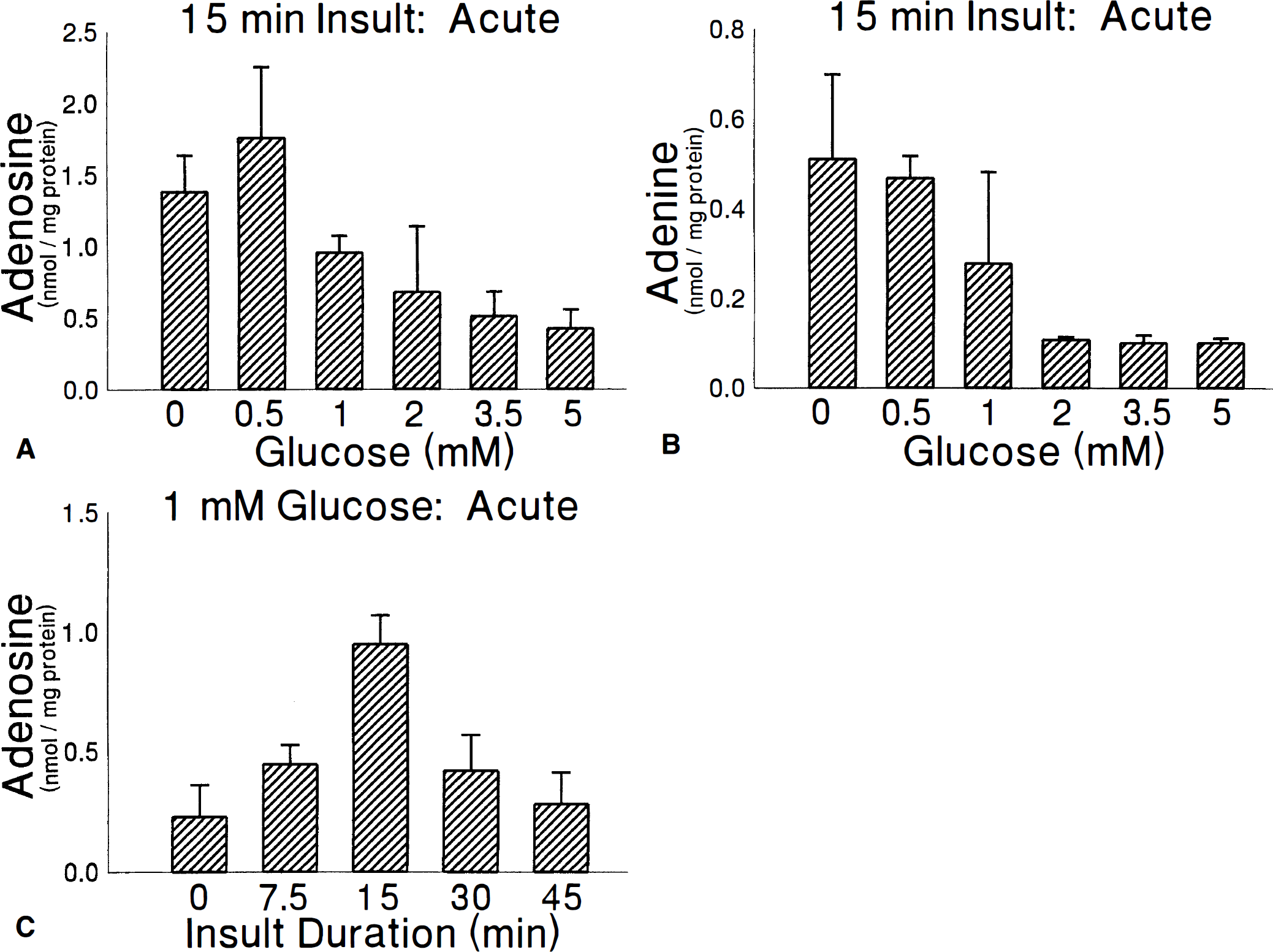
Influence of glucose on slice adenosine
The adenine nucleotide profile in slices exposed to air—5% CO2 is also highly dependent on the duration of insult (P < .001). There are significant decreases in ATP, ADP, and TAN with 1 mmol/L glucose beginning as early as 7.5 minutes and increasing with longer insult duration (Fig. 5B). ATP appears to stabilize by 30 minutes at less than 3 nmol/mg protein. There are no significant effects of insult duration on AMP or AEC between 7.5 and 45 minutes. AEC decreases to the target range of 0.4 to 0.5 over the first 15 minutes and is then stable thereafter (Fig. 3). Adenosine increases during the first 15 minutes of insult to 1.76±0.50 nmol/mg protein (Fig. 6A) and then gradually returns to pre-insult levels of 0.26±0.06 nmol/mg protein after 45 minutes of insult (Fig. 6C). The same effect occurs when slices are exposed to air—5% CO2 with 0.5 mmol/L glucose.
Several of the acute effects on adenine nucleotides persist when slices are returned to control conditions for 2 hours of reperfusion. There is a strong influence of glucose during the 15 minutes insult on slice ATP after 2 hours (Fig. 7A) but no longer any influence of insult glucose concentration on AMP or AEC. Neither is there any effect of glucose during insult on adenosine, inosine, or hypoxanthine, with the mean adenosine, in all groups 2 hours after return to control conditions, of 0.17±.09 nmol/mg protein. The influence of insult duration remains significant for ATP, AMP, AEC, and TAN even after 2 hours of reperfusion (Fig. 7B). By 30 minutes of insult, slices do not recover to a normal AEC after returning to control conditions for 2 hours. There is little further change as the insult increases from 30 to 45 minutes. Although there are significant differences in ATP, AMP, and AEC between the end of insult and recovery at 60 and 120 minutes (P < .001), there is no effect on TAN (P = .393), suggesting that most total adenine nucleotide loss occurs during the insult and also that there is little spontaneous repletion of TAN. This observation does not preclude an active cycling between adenine nucleotides, nucleosides, and purines, however.
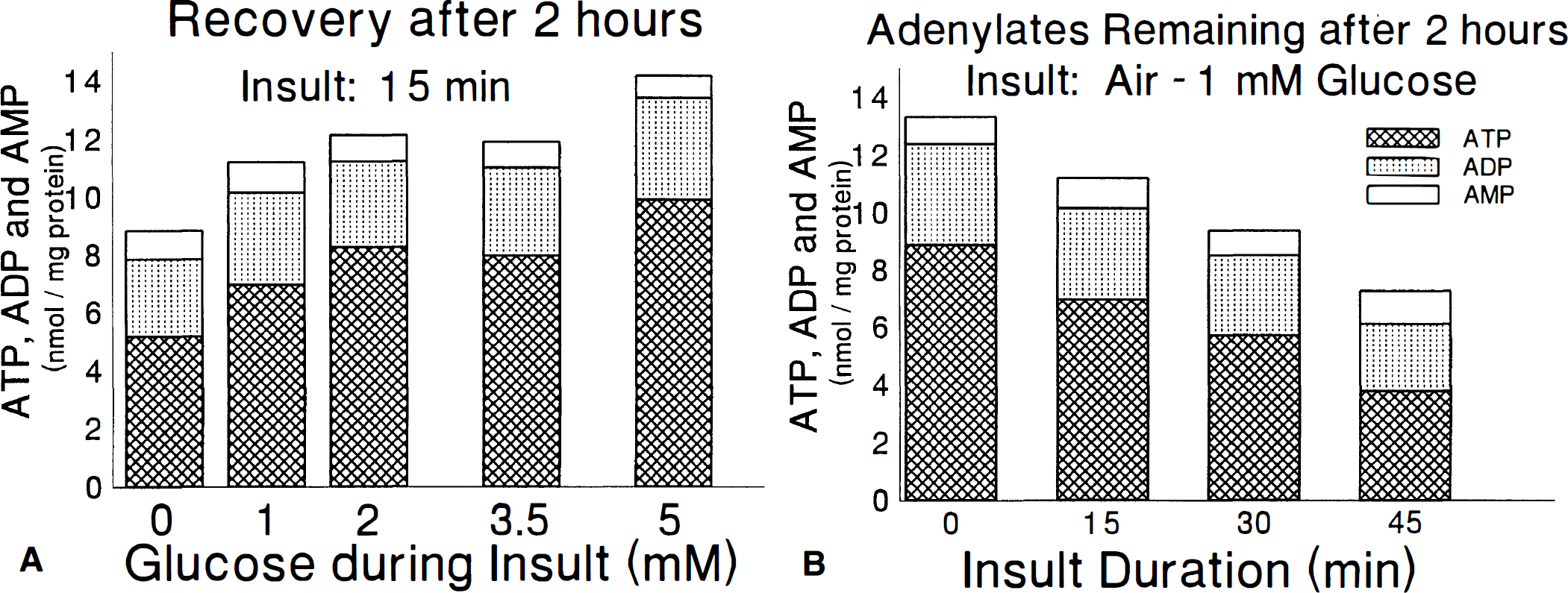
Measurements of adenine nucleotides in slices exposed to 95% air—5% CO2 and then returned to 95% O2—5% CO2 with 5 mmol/L glucose for 2 hours before freezing. The influence of varying the glucose concentration during a 15-minute insult
Histologic injury is readily apparent in slices exposed to air—50% Co2 with 1 mmol/L glucose and then to control conditions for 2 hours. The injury is limited to the CA1 and dentate regions (Fig. 8) with virtually no change in the injury score in CA2 (mean for all conditions is 2.8±.5), CA3 (3.0±.3) or CA4 (3.0±.4) whether the slices receive any insult or not. The degrees of injury in CA1 and dentate appear to remain stable as the insult duration is increased from 15 to 45 minutes.
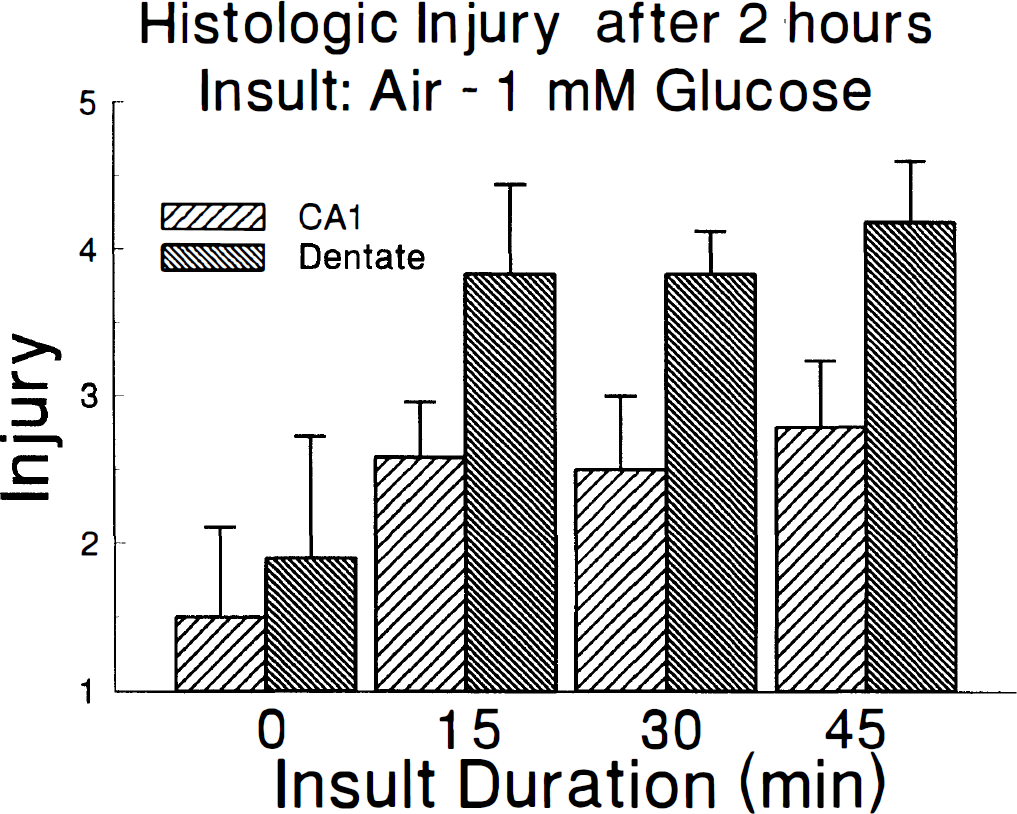
Histologic injury in CA1 and dentate that results when slices are exposed to 95% air—5% CO2 with 1 mmol/L glucose for varying times and then returned to 95% O2—5% CO2 with 5 mmol/L glucose for 2 hours. The insult in regions CA2, CA3, and CA4 were unchanged as duration of insult was varied.
These results suggest that a realistic and practical hippocampal brain slice model of reperfusion can be generated by exposing brain slices to air—5% CO2 with 1 mmol/L glucose for 30 minutes and then restoring the slices to control conditions for 2 hours. The slices reach and sustain an AEC of 0.4 to 0.5 for most of the insult and show both reduced TAN and observable histologic injury by the end of the reperfusion period. The moderate reductions in O2 and glucose are consistent with in vivo observations in the ischemic core (Ginsberg et al., 1977; Harris et al., 1987; Heiss et al., 1992). The peak of tissue adenosine at 15 minutes followed by a decrease at longer insults (Fig. 6C) is also a characteristic in vivo observation (Morimoto et al., 1991; Matsumoto et al., 1993; Phillis et al, 1994). By using 1 mmol/L glucose, potential metabolic disturbances with 0 mmol/L glucose are avoided but glucose is sufficiently low to permit study of alternative substrates for energy metabolism. The 30-minute insult is sufficiently long to study metabolism during the insult, if desired, and the injury after reperfusion should be insensitive to small variations in insult duration between 30 and 45 minutes.
Restoration of adenine nucleotides
The results on tissue adenine nucleotides of including 1 mmol/L AMP, adenosine or adenine as a treatment throughout the period of reperfusion after this insult are summarized in Fig. 9A. MANOVA reveals highly significant (P < .001) effects of treatment for all measures. Univariate analysis reveals similarly high significance for nearly all measures. Inspection of the results in Figure 9A reveals the expected effects of the 30-minute insult (time = 0) with reduced ATP, AEC, and TAN and increased AMP compared to the true controls but with no difference in ADP. It is also apparent that ATP, ADP, and TAN are increased by all three treatments relative to slices that received insult with reperfusion under control conditions. Tissue AMP is higher when slices are reperfused in the presence of AMP, but it does not differ from controls when slices are treated with adenosine or adenine. As a result, the highest values of AEC after 2 hours of reperfusion are seen with adenosine (0.79±0.34) and adenine (0.80±0.35). There are no differences among the three treatment groups as even the increase in tissue AMP during treatment with AMP does not reach statistical significance after correction for calculated extracellular AMP. There is no significant effect of recovery time between 1 and 2 hours on any measure and there are no significant interactions between treatment and recovery time.
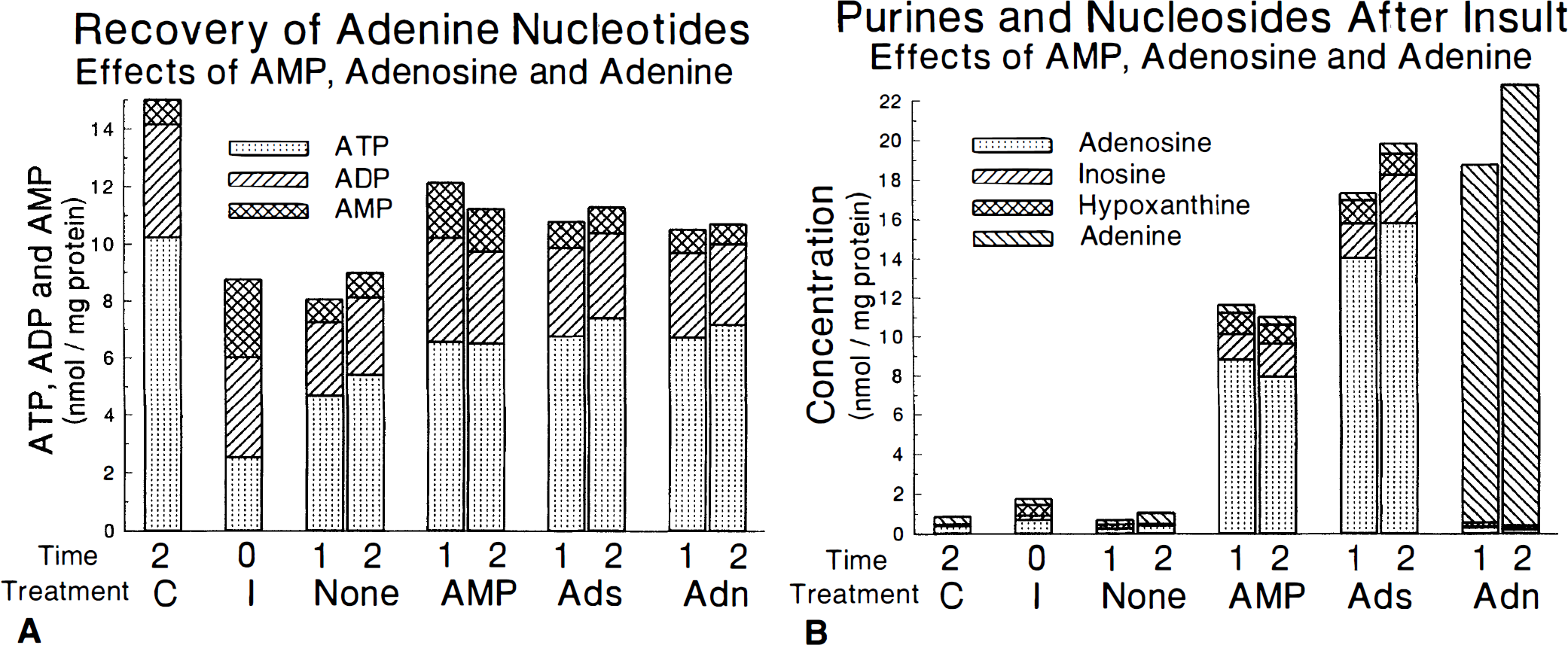
Time course of adenine nucleotides
Analogous results of treatments on tissue levels of adenosine, inosine, hypoxanthine, and adenine are shown in Figure 9B. Inosine (0.22±0.09) and hypoxanthine (0.54±0.14) increase acutely after 30 minutes of insult, but adenosine and adenine do not. There are very large effects of treatment during recovery on all measures (P < .001). As expected, treatment with adenosine substantially increases adenosine as well as inosine and hypoxanthine while treatment with adenine increases only tissue adenine. However, it is somewhat surprising that the increases in adenosine, inosine, and hypoxanthine induced by treatment with AMP are more than half of those seen with adenosine treatment. Covariate analysis reveals that all of these differences remain significant even when AMP is treated as a covariate. However, when adenosine is the covariate, the results for inosine and hypoxanthine are no longer significant. Values of these compounds after treatment with adenine are no different than controls. There are no significant effects of recovery time between 1 and 2 hours or interactions between treatment and recovery time.
The histologic effects of treatment with AMP, adenosine, and adenine during the 2-hour recovery period after a 30-minute insult with air—5% CO2 and 1 mmol/L glucose are summarized in Figure 10. The injury induced by the insult is readily apparent (P < .001). In addition, it is clear that only adenosine provides significant histologic protection against the transient hypoxic insult. Both parametric and nonparametric tests reveal that adenosine improves histology of all regions (P < .005) but CA2. Although there appears to be a trend toward improvement with AMP and adenine treatments, there is no significant benefit on histology despite the fact that these drugs increase adenine nucleotides to the same extent as adenosine.
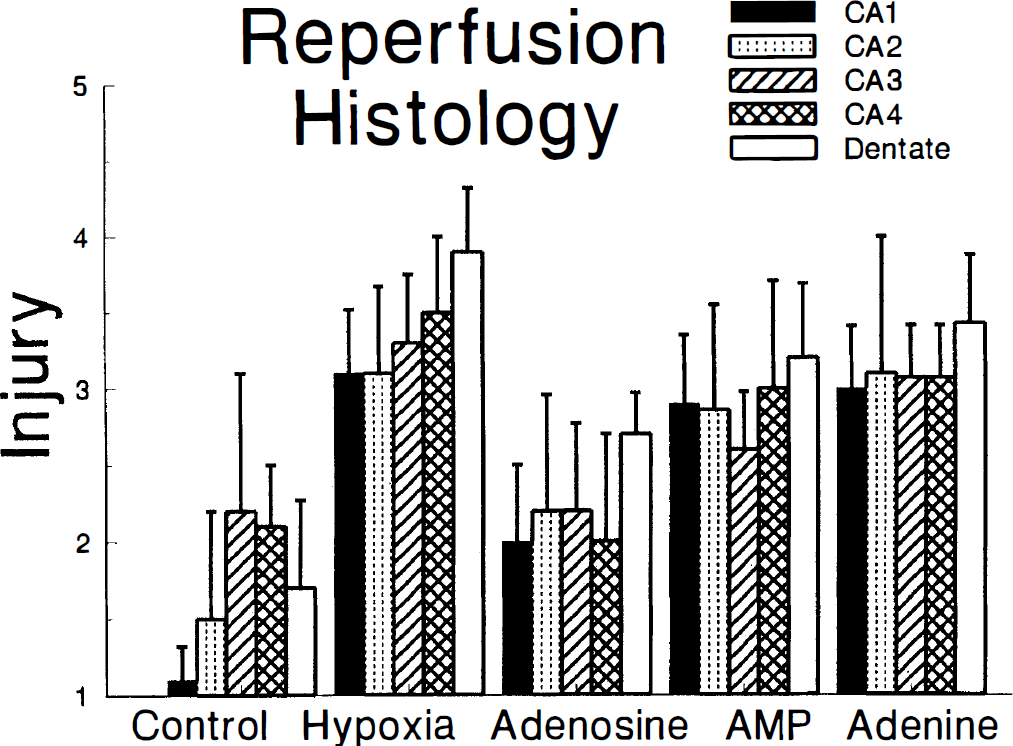
Influence of adenosine, AMP, and adenine on slice histologic injury. Control indicates slices incubated continuously in 95% O2—5% CO2 with 5 mmol/L glucose. Hypoxia indicates slices exposed to 95% air—5% CO2 with 1 mmol/L glucose for 30 minutes and then returned to 95% O2—5% CO2 with 5 mmol/L glucose for 2 hours with no buffer supplements. The remaining slices were exposed to the same insult and then for 2 hours to buffer equilibrated with 95% O2—5% CO2 with 5 mmol/L glucose, supplemented with 1 mmol/L adenosine, AMP, or adenine.
DISCUSSION
Revascularization with intravenous or intra-arterial thrombolytic agents after focal arterial occlusion presents new opportunities and challenges. Some of the emerging challenges include the need to reduce the risk of intracerebral hemorrhage, overcome the no-reflow phenomena, reduce reperfusion injury, and restore adenosine nucleotides to their pre-ischemic levels (Hallenbeck and Dutka, 1990). While the two former goals are best addressed with in vivo ischemia models or in vitro vascular preparations, the latter two goals may be greatly aided by the availability of a realistic in vitro model of ischemic cerebral metabolism. The present studies were intended to develop such a realistic brain slice model and to use the model to address specific questions regarding the best manner of restoring adenosine nucleotides during reperfusion after transient cerebral ischemia. Interpretation of cerebroprotective effects with this model is greatly simplified because brain slices lack CBF. Thus, any protection that is observed must be related to direct effects on neurons, glia, or ECS.
The brain slice model that uses transient anoxia with or without aglycemia has been among the most widely used and most successful models of transient ischemia (Whittingham et al., 1984; Schurr et al., 1986; Fowler, 1993; Pedata et al., 1993). There are, however, several disadvantages to this model, primarily related to the severity of the insult. In agreement with prior studies (Whittingham et al., 1984), our studies show that exposing slices to anoxia results in extremely low AEC within minutes of onset. Aglycemia worsens the severity of the insult still further. The value of AEC approaches 0.2 in this slice model, well below the value of 0.4 which has been observed in the core after focal arterial occlusion (Komatsumoto et al., 1987; Wagner et al., 1992; Folbergrova et al., 1995). In addition, the insult must be narrowly timed between 5 and 7 minutes because insults of less than 5 minutes produce little or no injury while insults longer than 7 minutes produce uniformly severe injury without recovery. Not only is this duration of insult at variance with the usual in vivo conditions of focal ischemia, it also reduces the tolerance for experimental error during the brain slice experiment and introduces the undesirable tendency of this insult to produce an “all or none” type of injury that is observed both physiologically and histologically (Fig. 4B; Taylor et al., 1985; Schurr et al., 1986; Weber and Taylor, 1994). These negative attributes provided the impetus to search for a brain slice model without these disadvantages. The desired model would achieve a stable AEC of 0.4, just as in vivo, and produce an easily observable graded injury after 2 hours which would be at once severe enough to be improved by cerebroprotective therapies but mild enough to be worsened by additional insult. By extending the insult duration to 30 minutes, not only is the experimental tolerance for variation in insult duration greatly increased, but there is now adequate time to conduct radiolabeling studies should that be desirable. The obvious first attempt at solution of this problem is to vary the degree of hypoxia and hypoglycemia and the duration of the insult until the desired properties are achieved. It quickly became apparent that 50% hypoxia is far too mild, achieving a stable AEC of only 0.7 between 30 and 150 minutes of insult. Reducing O2 to 40% produces an unstable AEC which decreases steadily from 0.6 after 30 minutes to 0.4 after 150 minutes. Also, because air—5% CO2 is a standard gas but 40% O2—5% CO2 is a specialty gas, the air mixture has a considerable cost advantage. Thus, subsequent experiments were based on an air—5% CO2 gas mixture. After considering a range of hypoglycemia and durations, an insult which consists of a 30-minute exposure to air—5% CO2 with 1 mmol/L seemed to approach the desired attributes most closely. This degree of hypoxia is similar to the 30-minute “moderately hypoxic” insult that has been shown to produce reversible electrophysiologic changes on reoxygenation of hippocampal brain slices (Schiff and Somjen, 1987) as well as increased release of adenosine and inosine (Fowler, 1993).
The diagram of metabolic pathways in Figure 1 does not show the full complexity of brain adenine nucleotide metabolism because the biochemical pathways are likely to be compartmentalized, with differences between neurons and glia and substantial metabolism occurring in the ECS as well (Fredholm, 1997). Under normal circumstances, ATP is released by neurons into the synaptic cleft and rapidly hydrolyzed to adenosine by a series of ectonucleotidases. Adenosine is also released directly. Extracellular adenosine plays an important role as a local neuromodulator and is one of the primary factors contributing to regulation of CBF (Van Wylen et al., 1986). At least three different adenosine transporters are active in brain to transport adenosine back into neurons or into glia (Parkinson et al., 1993). During resting conditions, most intracellular adenosine synthesis appears to derive from S-adenosylhomocysteine hydrolase. During ischemia, there is a large but transient increase in the release of adenosine as the higher concentration of AMP stimulates 5'-nucleotidase (Phillis et al., 1994; Latini et al., 1996). It is possible that the subsequent acidosis related to ischemia eventually inhibits 5'-nucleotidase (Bak and Ingwall, 1994). The higher levels of adenosine are sufficient to stimulate deamination to inosine and hypoxanthine, with possible value for glial protection (Haun et al., 1996). Excess release of inosine and hypoxanthine continues long after adenosine levels have begun to decrease (Morimoto et al., 1991; Matsumoto et al., 1993; Phillis et al., 1994). It also appears highly likely that purines generated during ischemia can be recycled to adenine nucleotides (Morimoto et al., 1991; Phillis et al., 1995) and, based on results in brain slices (Fredholm et al., 1994) and other tissues (Smolenski et al., 1991; Zoref-Shani et al., 1992; Bontemps et al., 1993), that this occurs via the purine salvage pathways for adenine and hypoxanthine.
Our results are consistent with this understanding and provide unambiguous evidence of the potential for cycling among adenine compounds in neural tissue, even in the absence of blood flow or functioning vasculature. Incubating slices with AMP or adenosine significantly increases the concentration of tissue adenine nucleotides (Fig. 9A). Consistent with the hypothesis of recycling, it is clear from Figure 9B that substantial amounts of AMP are converted to adenosine. In fact, slice adenosine accumulation after incubation with AMP is more than half as great as after incubation with adenosine itself. It is also clear that incubation of slices with adenine can significantly increase the concentration of ATP in slices, indicating the presence of considerable adenine phosphoribosyltransferase (PRT) activity, similar in degree to that of adenosine kinase. It is possible that adenosine kinase is more important at early stages of ischemia when adenosine is still high but that the capacity for adenine PRT may be important in recovering from severe ischemia when the breakdown of nucleosides increases purine levels. Presumably, hypoxanthine PRT plays a similar role. In addition, the fact that hypoxanthine did not covary with adenosine in our experiments while inosine did, suggests that degradation to purines may depend on something other than the mere presence of increased nucleoside levels. Although radiolabeling experiments would be necessary to provide direct evidence of incorporation of adenine into adenine nucleotides, in the absence of any significant benefit of adenine on tissue preservation, direct incorporation is the most likely possibility.
The histologic protection observed with adenosine is not present with adenine or AMP despite the fact that all three treatments increase adenine nucleotides to nearly the same extent. Thus, these results are consistent with other studies that have shown that the benefits of adenosine in hippocampus are mediated, at least in part, by stimulation of tissue adenosine receptors (Perez-Pinzon et al., 1996). Based on these results, exogenous adenosine should be considered further as a potential treatment for reducing ischemic injury. It is possible that exogenous administration would continue to stimulate adenosine receptors after the tissue ceases to produce large amounts of adenosine, so that the cerebroprotective effects known to occur with endogenous adenosine may be prolonged. Because adenine nucleotide metabolism appears to be heterogenous among brain cell types, a role for adenine and hypoxanthine PRT in cerebroprotection should not be excluded on the basis of this data. Incubation with adenine clearly increases ATP. Synthesis of AMP through adenine PRT appears to be slightly slower than through adenosine kinase because there is greater tissue accumulation of adenine than of adenosine with little accumulation of AMP. Nonetheless, resynthesis of ATP is equivalent and incubation with adenine produces the highest AEC of all treatments. In the absence of a reactive cerebral vasculature or of intact connections with other brain regions, the selectivity of adenosine for protection, despite equivalent metabolic effects of all three treatments, is most easily explained by postulating direct stimulation of hippocampal adenosine receptors on neurons and/or glia. Both A1 and A2 adenosine receptor subtypes have been identified on neurons in the CA1, CA3, and dentate regions of hippocampus (Cunha et al., 1994) and found to be functionally active with potential roles during cerebral ischemia (Nehlig et al., 1994: Von Lubitz et al., 1995). Finally, the failure of AMP to improve histology despite the increase in tissue adenosine may be explained if the protective effect of adenosine is extracellular but the adenosine derived from AMP is generated predominantly intracellularly, possibly related to re-uptake of AMP.
Salvaging brain tissue exposed to severe ischemia for more than a few hours poses one of the most difficult challenges in cerebral metabolism today. Successful strategies are likely to be complex and involve multiple factors. This study suggests that delivering high concentrations of adenosine to the reperfused tissue is of value both for increasing tissue adenine nucleotides and for maintaining stimulation of protective adenosine receptors after the endogenous levels of adenosine begin to decrease. Thus, adenosine should be considered in studies seeking to devise a “stroke cocktail” for local intra-arterial infusion after thrombolysis. Although adenine did not produce histologic improvement in the present studies, it was effective in restoring total tissue adenine nucleotides. Because adenine is less likely than adenosine to have systemic complications such as hypotension, it is still reasonable to continue studying adenine for cerebroprotection, perhaps in combination with adenosine receptor agonists.
Footnotes
Acknowledgment
The authors thank Dr. Clifford S. Patlak for his helpful discussions and for reading portions of the manuscript.