
Abstract
The effect of the glutamate analogue L-α-aminoadipate (αAA) on the release of glutamate and γ-aminobutyric acid (GABA) from rat hippocampal slices was investigated in vitro. Oxygen/glucose deprivation caused a large release of glutamate and GABA. αAA added during energy deprivation reduced the glutamate release in a dose-dependent manner (56% reduction at 5 mM), whereas GABA release was unchanged. We speculate that ischemic glutamate release from the brain is mediated by a low affinity transport mechanism that is blocked by αAA.
Brain cells maintain steep gradients of amino acids across the plasma membrane by means of specific carrier systems (Nicholls and Attwell, 1990). High affinity uptake of glutamate and γ-aminobutyric acid (GABA) is relatively well characterized, and several of the carrier proteins involved have been cloned (Guastella et al., 1990; Kanai and Hediger, 1992; Pines et al., 1992). Low affinity glutamate transport in brain tissue has been shown (Logan and Snyder, 1971), but the function of these carriers of has been less studied (Nicholls and Attwell, 1990).
The glutamate efflux that occurs during energy deprivation of the brain, has been accounted for by different mechanisms. A calcium-dependent vesicular release may play a role in the early and late stages of cerebral ischemia (Katayama et al., 1991), but the largest part of glutamate is probably released by calcium-independent, nonvesicular mechanisms (Ikeda et al., 1989; Hegstad et al., 1993). The major shift of ions across the plasma membrane that takes place (Hansen and Zeuthen, 1981) may result in the reversal of high-affinity carriers (Nicholls and Attwell, 1990), or activation of the mechanisms that operate during osmotic stress (Kimelberg et al., 1990).
In this article we show that L-α-aminoadipate, a glutamate analogue with several different effects, reduces the ischemic release of glutamate from rat brain.
METHODS
Hippocampal slices were prepared as described previously (Langmoen and Andersen, 1981). In brief, male Wistar rats (100–200 g) were decapitated, the brains were cooled for 2 min in artificial cerebrospinal fluid (ACSF) at 4°C, and the hippocampus was carefully dissected into 0.5 mm thick transverse slices. The slices were then incubated in ACSF saturated by a gas mixture containing 95% O2 and 5% CO2 for a 60–90 min rest period. The ACSF consisted of (in mM) NaCl 123; KCl 3.75; KH2PO4 1.25; MgSO2 1; CaCl2 2; NaHCO3 26; glucose (dextro-rotated) 10. The pH was 7.4.
The slices were then placed on transportable grids submerged in wells containing ACSF (35°C). After 30 min, the grids supporting the slices were transferred to five different test wells containing (1) control conditions: ACSF saturated with 95% O2/5% CO2 (test well A—control); (2) ACSF with 0 mM glucose, and oxygen substituted by 95% N2 (energy deprivation, [ED])(test well B); (3) ACSF with 0 mM glucose/95% N2, and 0.2 mM L-α-aminoadipate added (ED/αAA 0.2 mM)(test well C); (4) ED/αAA 1 mM (test well D); and (5) ED/αAA 5 mM (test well E). The slices were kept in these test wells for 20 min, then removed for determination protein content. ACSF samples were collected from the wells for amino acid determination, cleaned, and deproteinized by ultracentrifugation through a 5 × 103 Dalton cut-off filter (Nihon Millipore, Yonezawa, Japan), and stored at −70°C until analysis.
The content of glutamate and GABA in the ACSF was determined by high performance liquid chromatography (HPLC, Perkin Elmer, Norwalk, CT, U.S.A) after precolumn derivation with o-phthalaldehyde (Fluka Chemie AG, Buchs, Switzerland)/3-mercapto propionic acid (Sigma, St. Louis, MO, U.S.A) at pH 10.5 (Haugstad et al., 1995). Fluorescence peaks (Perkin Elmer LC 240, excitation wavelength 340 nm, emission wavelength 460 nm) were integrated by Perkin Elmer 1020X software.
The protein content of the slices was determined according to the method of Lowry (Lowry et al., 1951). The results were tested statistically using the Wilcoxon rank sum test and are presented as mean ± standard deviation.
RESULTS
During control conditions (test well A), the slices released 920 ± 320 pmol glutamate per mg protein (n = 7). Slices exposed to 20 min of combined oxygen and glucose deprivation (test well B) released 21,260 ± 6,880 pmol/mg (2,310% of control, p = 0.018). The slices exposed to 0.2 mM αAA during energy deprivation (test well C), released 16,320 ± 4,320 pmol/mg (76% of well B, p = 0.063). Those exposed to 1 mM αAA (well D) released 9,710 ± 1,730 pmol/mg (46% of well B, p = 0.028), and those exposed to 5 mM αAA (well E) released 9,350 ± 2,220 pmol/mg (44% of B, p = 0.018). Thus, αAA significantly reduced the release of glutamate during energy deprivation in a dose-dependent manner (Fig. 1).
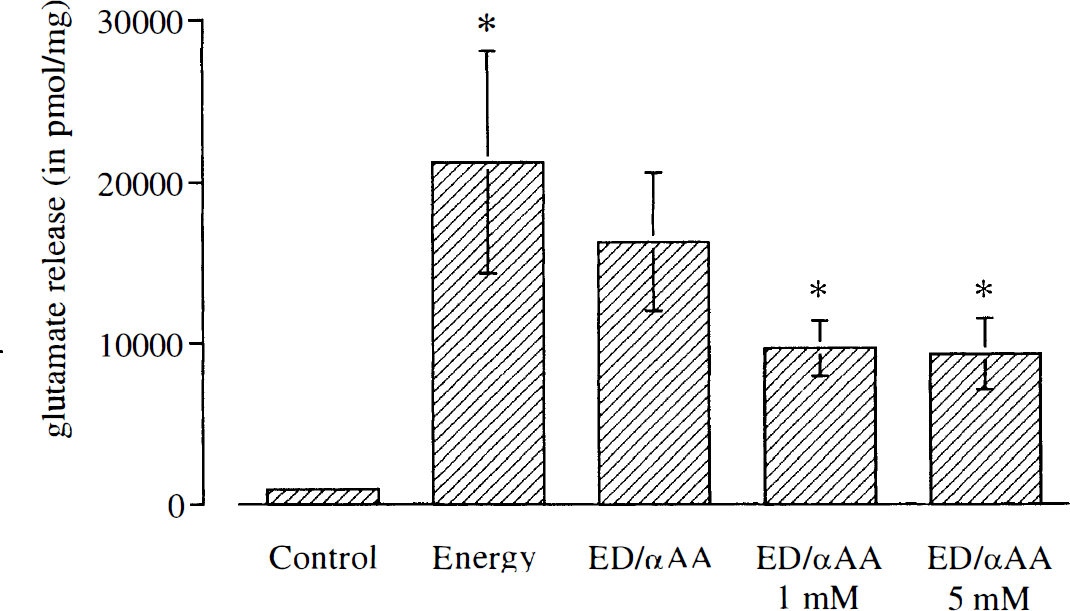
Glutamate release from rat hippocampal slices in five sets of test wells during 20 minutes. The wells contained A) Control conditions: ACSF saturated with 95% O2/5% CO2 (Control), B) Glucose free ACSF with oxygen substituted by 95% N2 (Energy Deprivation, ED) and C) ED with 0.2 mM L-α-aminoadipate added (ED/αAA 0.2 mM), D) ED/αAA 1 mM and E) ED/αAA 5 mM. Values represent mean ± standard deviation from 7 experiments. Asterisks denote p < 0.05 for comparison between B versus A, and D and E versus B, by the Wilcoxon rank sum test.
The GABA release during control conditions (A) was 420 ± 330 pmol/mg (n = 7). During energy deprivation (B) it increased to 8,060 ± 2,700 pmol/mg (1,920% of control, p = 0.018). Slices exposed to energy deprivation with 0.2 mM αAA added (C) released 7,060 ± 1,180 pmol/mg (88% of B), those exposed to ED with 1 mM αAA added (D) released 8590 ± 1720 pmol/mg (107% of B), and slices exposed to ED with 5 mM αAA present (E), released 8640 ± 3030 pmol/mg (107% of B, see Fig. 2). None of these concentrations of αAA significantly affected the GABA release from the slices exposed to energy deprivation.
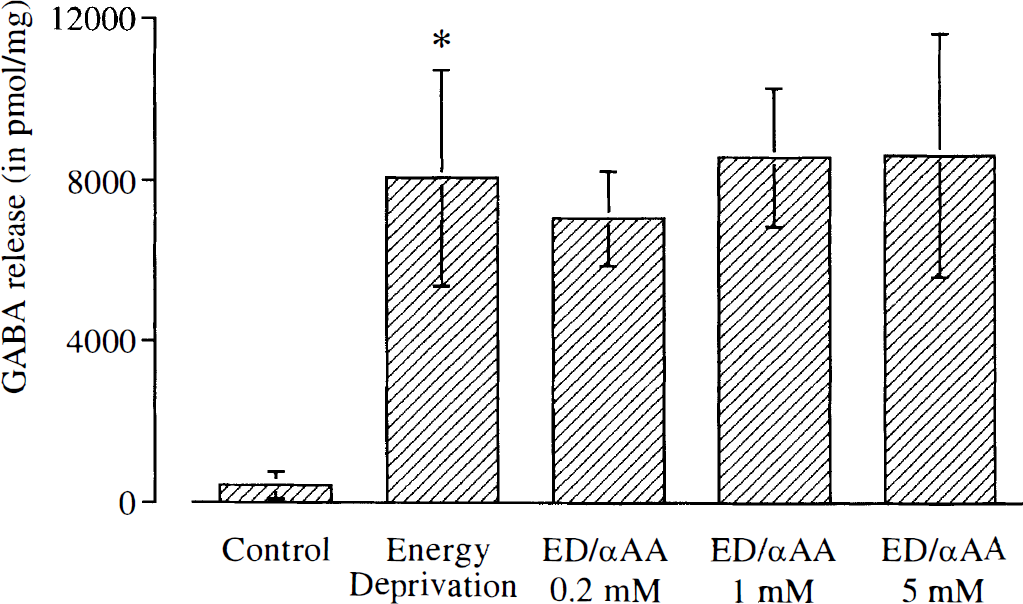
GABA release from rat hippocampal slices in three sets of test wells during 20 minutes. The wells contained A) Control conditions: ACSF saturated with 95% O2/5% CO2 (Control), B) Glucose free ACSF with oxygen substituted by 95% N2 (Energy Deprivation, ED) and C) ED with 0.2 mM L-α-aminoadipate added (ED/αAA 0.2 mM), D) ED/αAA 1 mM and E) ED/αAA 5 mM. Values represent mean ± standard deviation from 7 experiments. Asterisks denote p < 0.05 for comparison between B versus A by the Wilcoxon rank sum test.
DISCUSSION
Several uptake systems for glutamate and GABA have been described (Christensen, 1984; Danbolt, 1994). Sodium dependent high affinity transport of glutamate and GABA in the brain is well characterized (Guastella et al., 1990; Nicholls and Attwell, 1990; Kanai and Hediger, 1992; Pines et al., 1992). Low affinity carriers of amino acids have been extensively studied in peripheral cells (Christensen, 1984; Bannai, 1986; Miura et al., 1992) and in synaptosomal vesicles from the brain (Naito and Ueda, 1985; Fykse and Fonnum, 1988; Maycox et al., 1990). Low affinity uptake probably also participates in removing peak glutamate concentrations (≈1 mM) during synaptic transmission (Waniewski and Martin, 1984; Clements et al., 1992)
αAA has complex effects on brain cells. It inhibits high-affinity glutamate uptake when tested in synaptosomes (Robinson et al., 1991). Transport studies of the cloned glutamate carriers have shown that αAA inhibits the glial transporter GLT-1 (Pines et al., 1992) and the neuronal transporter EAAC (IC50 = 165 μM) (Kanai and Hediger, 1992), but not the GLAST-1 carrier (Klöckner et al., 1994). In addition, αAA inhibits the glutamate/cystine antiporter (Bannai, 1986; Miura et al., 1992; Kato et al., 1993). Further, it is a selective gliotoxin (Olney et al., 1971; Lund Karlsen, 1978). Recently, αAA has been shown to inhibit glutamate release from microglia evoked by bacterial toxins (Piani and Fontana, 1994). αAA is a potent inhibitor of low-affinity glutamate uptake into presynaptic vesicles (Fykse et al., 1992). It inhibits the glial enzymes glutamine synthetase and 7-glutamylcysteine synthetase (McBean, 1994). The D-form is also a potent antagonist at the NMDA receptor (Watkins et al., 1990; Clements et al., 1992).
The present study shows that αAA efficiently inhibits the release of glutamate evoked by energy deprivation from rat brain in a concentration range from 10–4 to 10–3 M, the same range that inhibits high affinity glutamate uptake in synaptosomes from rat cerebral cortex (Robinson et al., 1991). GABA release was unaffected. The results indicate that the two amino acids at least in part are liberated by separate mechanisms. We have shown earlier that (1) ischemic glutamate release occurs by mechanisms separate from osmotic release (Haugstad et al., 1995; Haugstad and Langmoen, 1996); 2) high-affinity uptake inhibitors increase ischemic glutamate efflux (Haugstad and Langmoen, 1995) by blocking uptake rather than increasing release (Haugstad, 1996); (3) high-affinity glutamate transport into glia cells continues during cerebral ischemia, while neuronal compartments are depleted (Aas et al., 1993; Hegstad et al., 1996); (4) 1 mM of L-methionine-S-sulphoximine (MSO), that inhibits the glial enzyme glutamine synthetase and γ-glutamylcysteine synthetase, does not alter ischemic glutamate release from brain slices (Haugstad and Langmoen, unpublished results). Thus, the dose-dependent effect of millimolar concentrations of the long-chained glutamate analogue αAA on ischemic glutamate efflux is probably mediated via mechanisms different from the effect on glial enzymes or high-affinity glutamate carriers. We speculate that low-affinity transport systems, which generally have a high transport rate compared with high-affinity transporters and the glutamate/cystine antiporter (Kato et al., 1993; Danbolt, 1994; Hegstad et al., 1995), may be involved in the ischemic release of glutamate from brain tissue. The results further point towards the possibility that ischemic release and the release evoked by inflammatory processes may share common mechanisms (Piani and Fontana, 1994).
Footnotes
Acknowledgments:
This work was supported by The Norwegian Council on Cardiovascular Diseases and The Lærdal foundation. We are grateful to Professor Ansgar Aasen, Institute for Surgical Research, and Professor Helge Nornes, Department of Neurosurgery, for providing ample facilities and support.