
Abstract
The in vivo and in vitro bindings of radiolabeled rotenoids to mitochondrial complex I of rat striatum were examined after unilateral intrastriatal injections of quinolinic acid or 1-methyl-4-phenylpyridinium salt (MPP+). Quinolinic acid produced significant, similar losses of in vivo binding of [11C]dihydrorotenol ([11C]DHROL: 40%) and in vitro binding of [3H]dihydrorotenone ([3H]DHR: 53%) in the injected striata at 13 days after the injection of neurotoxin. MPP+ reduced in vivo binding of [11C]DHROL (up to −55%) as measured 1.5 to 6 h after its administration. Reductions of in vivo [11C]DHROL binding after either quinolinic acid or MPP+ injections did not correlate with changes in striatal blood flow as measured with [14C]iodoantipyrine. These results are consistent with losses of complex I binding sites for radiolabeled rotenoids, produced using cell death (quinolinic acid) or direct competition for the binding site (MPP+). Appropriately radiolabeled rotenoids may be useful for in vivo imaging studies of changes of complex I in neurodegenerative diseases.
Although the underlying causes of degenerative neurological diseases, such as Huntington's, Alzheimer's, and Parkinson's diseases and amyotrophic lateral sclerosis, remain unknown, it has been hypothesized that they might result from an impairment of neuronal energy metabolism (for review; see Beal, 1995). The loss of ATP synthesis may produce prolonged depolarization, activation of voltage-dependent-N-methyl-D-aspartate (NMDA) receptors, and subsequent “weak excitotoxicity” (Albin and Greenamyre, 1992). Low levels of ATP might also compromise the ability of cells to sequester and store intracellular calcium. In Parkinson's disease, evidence for mitochondrial dysfunction includes the observation that 1-methyl-4-phenylpyridinium ion (MPP+), the active metabolite of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), is an in vitro and in vivo inhibitor of mitochondrial complex I activity (Nicklas et al., 1985; Ramsay et al., 1986a,b; Tipton and Singer, 1993). Although the findings are controversial, reduced levels of complex I enzymatic activity have been reported in tissues of parkinsonian patients (DiMauro, 1993; Schapira and DiMauro, 1994; Mann et al., 1994). It remains unresolved, however, whether abnormalities in complex I play a primary or secondary role in the inception or progression of neurodegeneration and whether these alterations are due to genetic defects in mitochondrial or nuclear DNA or are the result of an environmental toxin or oxidative stress (Jenner et al., 1992).
Evaluation of complex I changes in living patients at earlier stages of disease will require new methods for noninvasive measurement of energy metabolism that are selective for mitochondrial oxidative capacity. Positron emission tomography (PET) might be employed for in vivo study of mitochondrial enzyme complexes with the development of appropriate radiotracer methodology. In vitro binding studies with [3H]dihydrorotenone ([3H]DHR, 66.2 Ci/mmol) have shown the feasibility of this approach, with saturability and pharmacology consistent with rotenoid binding to complex I (Greenamyre et al., 1992). Thus, in contrast to earlier attempts to use very low-specific-activity [14C]rotenone (2.36 mCi/mmol) as an in vitro radioligand (Horgan et al., 1968), the availability of a high-specific-activity tritiated rotenoid has allowed demonstration of changes in complex I binding sites for this radioligand following selective rat brain lesions (Blandini et al., 1995).
Extension of these encouraging in vitro results to in vivo imaging of human disease requires development of rotenoids appropriately labeled with gamma-emitting radionuclides. We recently reported the synthesis of two positron-emitter-labeled rotenoids, [11C]rotenone ([11C]ROT) and [11C]dihydrorotenol ([11C]DHROL), as potential in vivo radiotracers for imaging of complex I using PET (Charalambous et al., 1995a, 1995b). Both radiotracers incorporate carbon-11 (half-life = 20.4 min) in the 2-methoxy substituent of the aromatic A-ring (Fig. 1) and can be prepared in high specific activities (>500 Ci/mmol). [11C]DHROL, a rotenone derivative incorporating minor structural modifications that should not alter greatly the in vitro affinity for complex I, was developed as a more hydrophilic and metabolically stable radiotracer. Regional brain distribution studies in mice and rats showed excellent brain uptake and widespread distributions of both [11C]ROT and [11C]DHROL in all areas examined (Charalambous et al., 1995b), consistent with the distribution of [3H]DHR bnding found in vitro (Greenamyre et al., 1992). The further development of [11C]rotenoids for human brain imaging of complex I requires a demonstration that, in vitro and in vivo, such radioligands bind to this mitochondrial enzyme. Determination of in vitro binding affinites of new rotenoids is relatively straightforward using the recently developed in vitro assay for [3H]DHR binding (Greenamyre et al., 1992; Blandini and Greenamyre, 1995a, b ). In vivo, however, demonstration of specific binding to complex I is more challenging: in contrast to radioligand development for neurotransmitter receptors and transporters, where in vivo pharmacological specificity can be demonstrated by pretreatment of animals with sufficient amounts of competing ligands to occupy essentially all of the expected binding sites, it is not possible to inhibit totally whole brain complex I because of the toxicity of these enzyme inhibitors. We thus took two different approaches to demonstrate the in vivo specificity of radiolabeled rotenoids. First, because mitochondria are enriched in neurons versus glia, unilateral quinolinic acid (QA) lesions of the rat striatum were used to model the loss of mitochondrial binding sites resulting from neuronal death. Second, rotenoid binding in vivo was assessed shortly after unilateral striatal injections of MPP+ as a means to study direct competition for the binding site by this complex I inhibitor. The results of these experiments suggest that labeled rotenoids may have potential as in vivo radioligands for the study of complex I losses in diseased states.
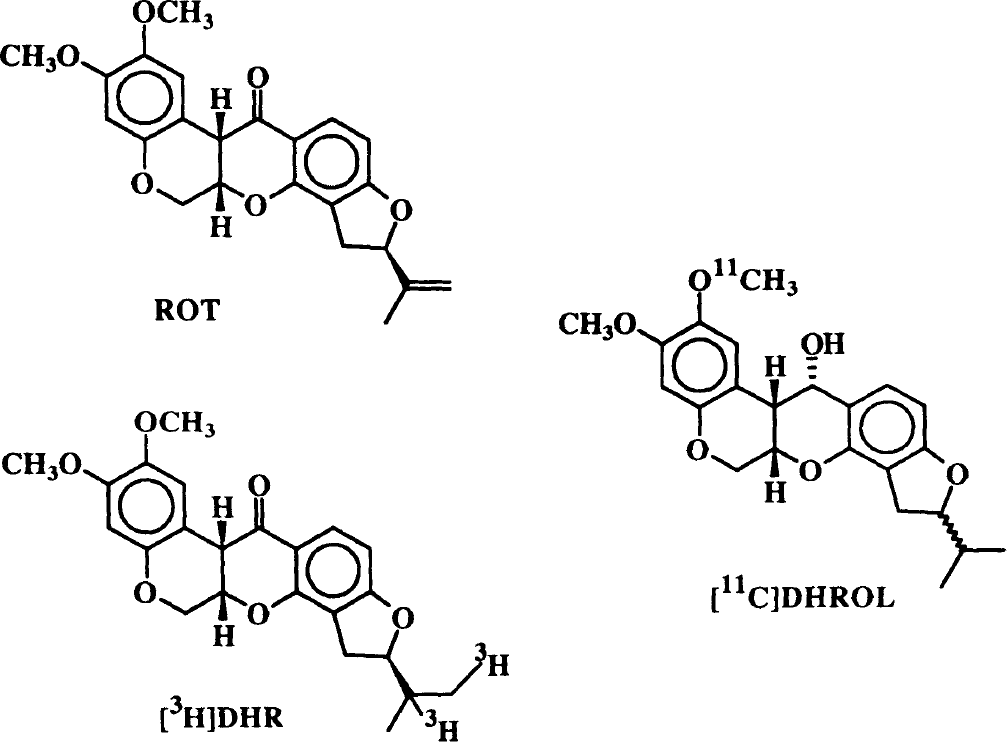
Structures of rotenone (ROT) and the two radioligands used in this study, [3H]dihydrorotenone ([3H]DHR) and [11C]dihydrorotenol ([11C]DHROL).
MATERIALS AND METHODS
Chemicals and drugs
Purchased chemicals include ketamine (Warner Lambert Co.), xylazine (Mobay Co.), MPP+ (iodide salt, Research Biochemicals International), and [14C]iodoantipyrine ([14C]IAP; DuPont). [11C]DHROL (>500 Ci/mmol) was synthesized according to a published procedure (Charalambous et al., 1995b) and was formulated in 10% ethanol in saline. [3H]Dihydrorotenone (DHR, 66.2 Ci/mmol) was custom synthesized and purified by Amersham as described previously (Greenamyre et al., 1992). All other reagents were obtained from Sigma Chemicals.
In vitro [3H]dihydrorotenone binding and determination of IC50s for dihydrorotenol and rotenone
Slide-mounted brain sections were incubated with 10 nM [3H]DHR in 50 mM Tris-HCl buffer (pH 7.6) containing 1% bovine serum albumin for 120 min at 20°C. Dihydrorotenone is insoluble in aqueous solutions and was added to the assay in a minimal volume of dimethyl sulfoxide (DMSO) not exceeding a final DMSO concentration of 1% (vol/vol). Nonspecific binding was defined by 100 μM unlabeled rotenone. Under these conditions, 80 to 90% of binding was specific. After incubation with [3H]DHR, the rinse consisted of 60 min in assay buffer, followed by two 10-min rinses in 50 mM Tris-HCl and a brief dip in distilled H2O. The slides were dried under a stream of warm air, mounted in X-ray cassettes, apposed to tritium-sensitive film with radioactive standards, and exposed for 2 to 4 weeks. Autoradiograms were analyzed using a video-based image analysis system (MCID, Imaging Research, St. Catharines, Ontario, Canada). DHROL and ROT competition for specific [3H]DHR bnding were assessed over the concentration range of 1 nM to 1 mM (n = 4). Data were analyzed with the iterative nonlinear curve fitting program LIGAND (Munson and Rodbard, 1980).
General method: unilateral intrastriatal injections
All animal protocols were approved by the University of Michigan Committee on the Use and Care of Animals. Studies were done in male Sprague-Dawley rats (150–200 g, Charles River). Animals were anesthetized with ketamine (85 mg/kg, i.m.) and xylazine (15 mg/kg). They then were injected stereotactically in the right striatum with phosphate-buffered saline solutions of either quinolinic acid (150 nmol, 1 μl), MPP+ iodide (100 nmoles, 1 μl), or isotonic saline (pH = 7.4, 1 μl) over 4 min at the following coordinates: 1.0 mm anterior to bregma, 2.6 mm lateral to midline, and 5.0 mm below dura. Studies of radiotracer binding were done at 13 days (QA) or 1.5 to 6 h (MPP+) after neurotoxin injection as described in the following section.
General method: in vivo striatal uptake of radiotracers
Animals were anesthetized and a catheter inserted in the right femoral vein. For the QA lesion studies, animals were studied in the awake but restrained state. For the MPP+ studies, animals were kept under anesthesia (ketamine, 85 mg/kg i.m. every hour) for the entire procedure. Each animal was first injected with approximately 500 μCi of [11C]DHROL, and after 29.5 min a second injection of 5 μCi [14C]iodoantipyrine ([14C]IAP) was made. Thirty seconds later, the animals were killed by decapitation. The brains then were rapidly removed and the left and right striata and cerebellum dissected. The tissues were quickly counted for 11C in a Packard 5780 Auto-gamma counter and then weighed. Following solubilization with 100 ml Solvable for 3 days, 100 ml glacial acetic acid and 1 ml Universal Scintillation Fluid were added, and the samples were counted for 14C in a Packard 4530 Liquid Scintillation counter. For each carbon isotope, data were calculated as the percent of injected dose per gram of body weight. Radioactivity concentrations in the injected sides were compared with those in the uninjected sides and are expressed as asymmetry ratios (ratios of radioactivity uptake in the right versus the left striata).
In vitro [3H]dihydrorotenone binding in QA-lesioned animals
A subset of the QA-lesioned rats was studied using in vitro [3H]DHR autoradiography. The rats (n = 6) were anesthetized, and decapitated, and the brains were rapidly removed and frozen at −800°C until being cut in a cryostat. Slide-mounted brain sections were stored at −800°C until assayed for [3H]DHR binding as described above. Specific binding was measured in both striata (QA-lesioned and control) from each animals, and the results are expressed as right/left asymmetries.
Statistical analyses
Side-to-side differences in radiotracer distributions were analyzed using a paired Student's t test. The statistical significance of asymmetry ratios between treated and control groups was assessed using an unpaired Student's t test. A p value < 0.05 was considered significant.
RESULTS
Dihydrorotenol inhibition of [3H]dihydrorotenone binding
As described previously, rotenone potently inhibited in vitro [3H]DHR binding with inhibiting concentration (IC50s) ranging from 8.2 ± 1.3 nM in cerebellar molecular layer to 19.9 ± 5.0 nM in striatum (data not shown). Dihydrorotenol was less potent, with IC50 values ranging from 77 ± 21 nM in deep layers of cerebral cortex to 113 ± 8 nM in the cerebellar molecular layer (Fig. 2). Hill coefficients were not significantly different than 1, consistent with a single high-affinity binding site for both rotenoids.
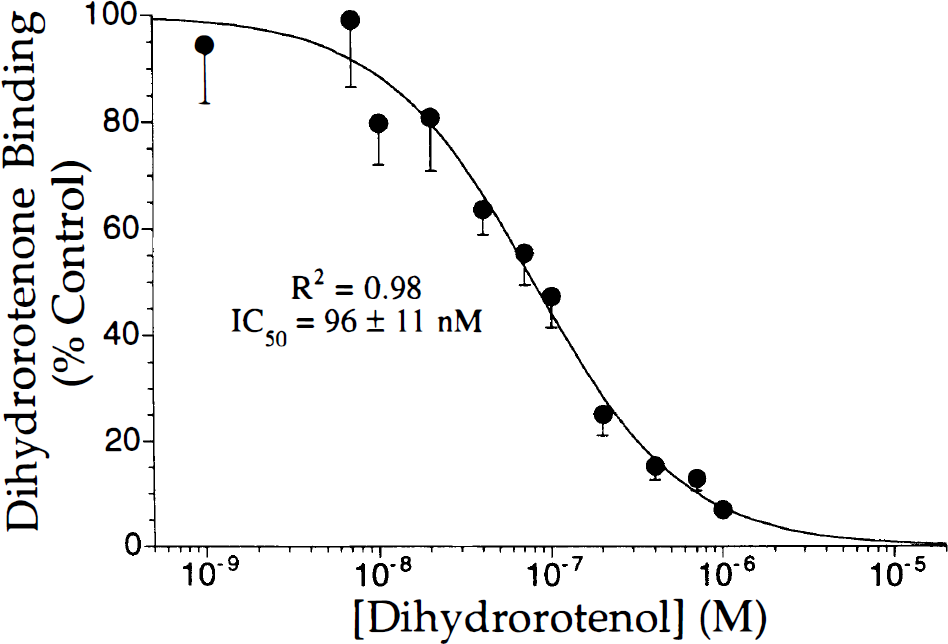
Inhibition by dihydrorotenol (DHROL) of [3H]dihydrorotenone ([3H]DHR binding to the superficial layers of the cerebral cortex of rat brain. Similar IC50s were obtained in striatum and other brain regions. Hill slopes did not differ significantly from 1. Points represent mean ± SD; n = 4.
In vivo striatal [11C]dihydrorotenol uptake after quinolinic acid lesions
In control animals injected with saline, there were no statistical differences between in vivo [11C]-DHROL accumulation in right versus left striata (asymmetry index = 1.002 ± 0.07, p = 0.98).
Thirteen days after QA injection, in vivo striatal accumulations of [11C]DHROL on the injected side were significantly reduced in each animal (p = 0.001, right vs. left striata) (Fig. 3). The asymmetric [11C]-DHROL retention in QA-lesioned animals (right/left = 0.61 ± 0.08) was significantly different (p < 0.005) from the asymmetry in saline-injected animals. In the QA-injected animals, there was also reduced [14C]IAP uptake in the injected striatum in five of six rats: For those five animals, the reductions were statistically significant (right vs. left; p < 0.05), but inclusion of the single animal with a right/left ratio > 1 made the asymmetry insignificant for the whole group. In saline-injected animals, there was no asymmetry in [14C]IAP uptake (p = 0.35). There was no significant difference in [14C]IAP uptake in the QA-lesioned group compared with the saline-treated animals (p = 0.24), but exclusion of the single animal QA-lesioned animal with the high right/left asymmetry produced significant differences between the groups (p < 0.001).
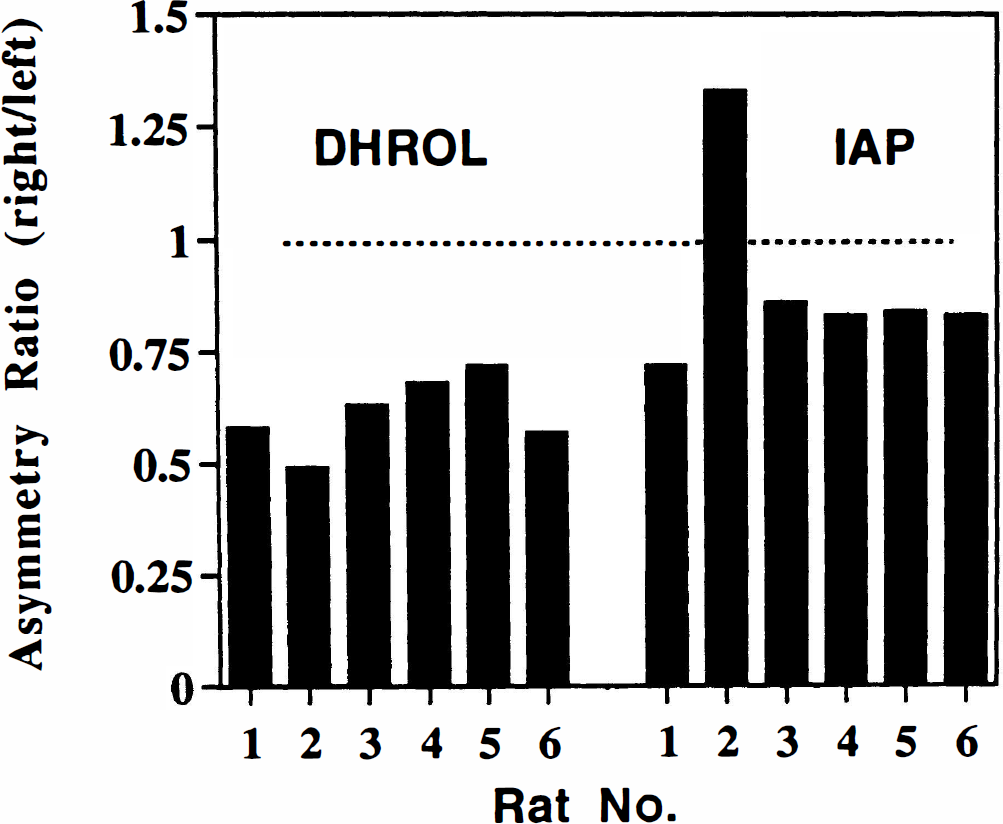
Asymmetry of striatal uptake of [11C]dihydrorotenol ([11C]DHROL) and [14C]iodoantipyrine ([14C]IAP) in rat brain following unilateral striatal injection of quinolinic acid. Data are shown as asymmetry ratios (% injected dose/gram tissue, right/left striatum) for both radiotracers in each animal. Average asymmetry ratios were [11C]DHROL, 0.61 ± 0.08; and [14C]IAP, 0.90 ± 0.21. The asymmetries in [11C]DHROL uptake were significantly (p < 0.05) greater than the asymmetries in [14C]IAP uptake.
In the QA-lesioned group, the asymmetry of [11C]DHROL uptake was consistently larger than the asymmetry seen in [14C]IAP uptake, and this difference was statistically significant with inclusion (p < 0.05) or exclusion (p < 0.005) of the animal with the [14C]IAP ratio > 1. For both control and QA-lesioned groups, there were no significant differences in striatal weights (right vs left or between groups).
In vitro striatal [3H]dihydrorotenone binding after quinolinic acid lesions
In QA-lesioned animals, there was a marked reduction in specific [3H]DHR binding in the injected striatum (Fig. 4). The lesion/control asymmetry was 0.47 ± 0.08 (p < 0.002).
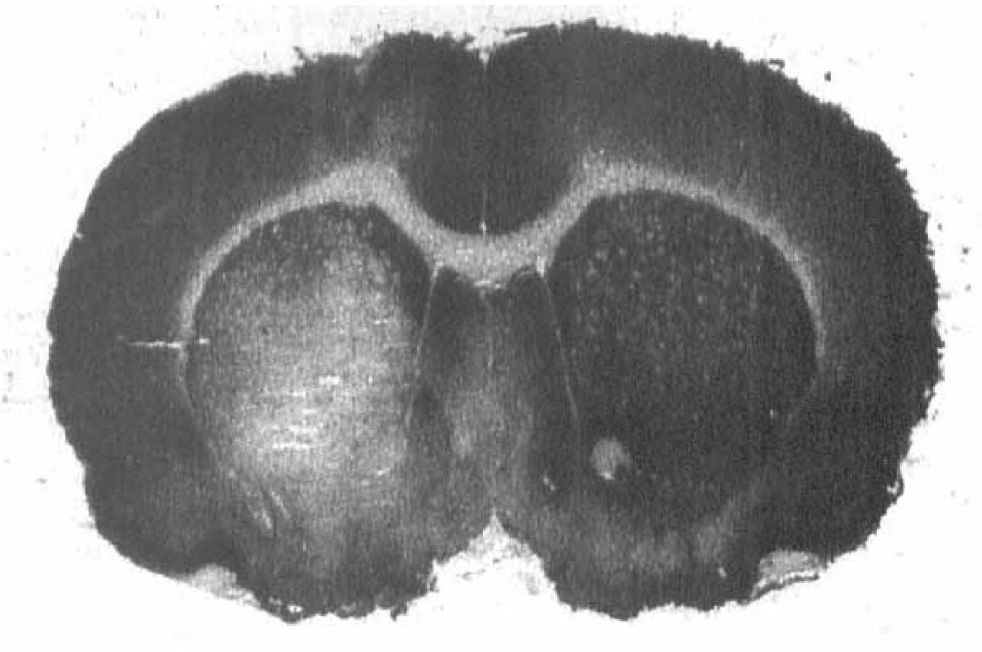
Autoradiogram of [3H]dihydrorotenone ([3H]DHR) binding to a section of brain from a rat that had received a quinolinate lesion 13 days previously. Note the marked decrease in binding in the region of the quinolinate lesion (right striatum, shown on left).
In vivo striatal [11C]dihydrorotenol uptake after MPP+ injections
Group A: controls. In the rats that received injections of saline in the right striatum 1.5 h before, no significant changes were observed for [11C]DHROL (mean asymmetry ratio, 1.00 ± 0.09) or blood flow ([14C]IAP, mean asymmetry ratio 0.99 ± 0.14) between the MPP+-injected and uninjected sides (individual data not shown).
Group B: average MPP+ exposure 1.5 hours. The effects of MPP+ at 1.5 h after injection on striatal concentrations of [14C]IAP and [11C]DHROL are shown in Fig. 5. The uptake of [11C]DHROL was variable and showed either unchanged or decreased (as much as 40%) radioactivity concentrations in the MPP+-injected sides (mean asymmetry ratio, 0.83 ± 0.07; p = 0.09, right vs. left). Conversely, there was a general increase (as much as 46%) in [14C] IAP uptake (mean asymmetry ratio, 1.22 ± 0.09; p = 0.57, right vs. left) in the MPP+-injected striata. Asymmetry in [11C]DHROL was significantly different from that for [14C]IAP (p < 0.001).
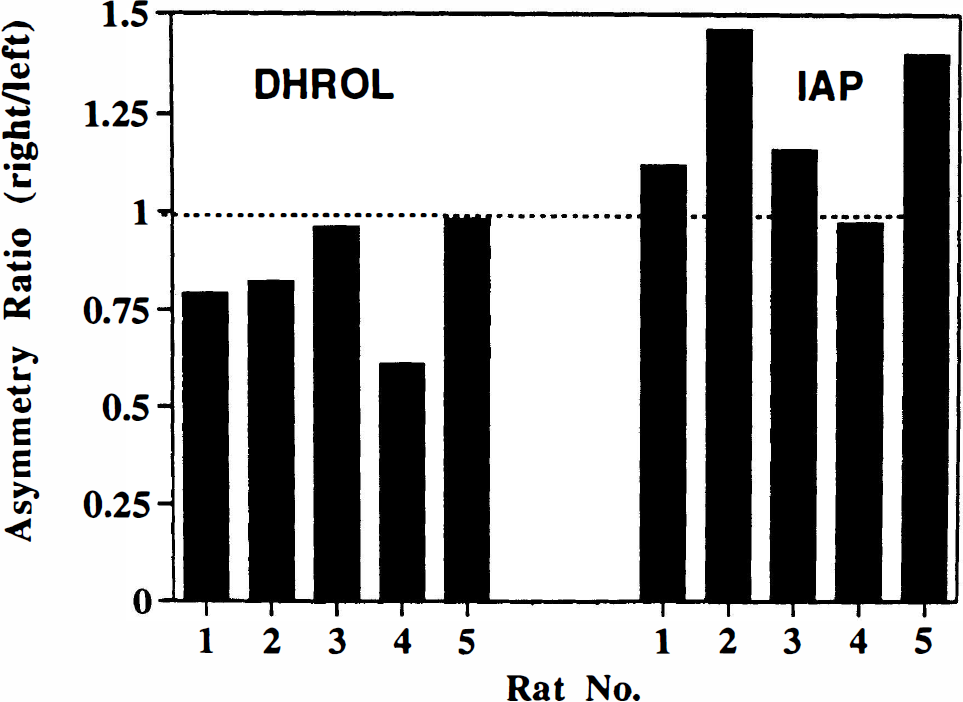
Asymmetry of striatal uptake of [11C]dihydrorotenol ([11C]DHROL) and [14C]iodoantipyrine ([14C]IAP) in the rat brain 1.5 h after unilateral striatal injection of 100 nmol MPP+. Data are shown as asymmetry ratios (% injected dose/g tissue, right/left striatum) for both radiotracers in each animal. Average asymmetry ratios were [11C]DHROL, 0.83 ± 0.07; and [14C]IAP, 1.22 ± 0.09. The asymmetries in [11C]DHROL were significantly different (p < 0.001) from the asymmetries in [14C]IAP uptake.
Group C: average MPP+ exposure 6 h. The effects of MPP+ on the concentrations of [14C]IAP and [11C]DHROL in the striatum at the later time point are shown in Fig. 6. For [11C]DHROL, the radioactivity concentrations were consistently lower (up to 55%) in the MPP+ injected side (mean asymmetry ratio, 0.66 ± 0.05: p = 0.005, right vs. left). In the uptake of [14C]IAP, there were highly variable changes in the MPP+-injected striata (mean asymmetry ratio 0.98 ± 0.11; p = 0.86 right vs. left). Linear regression analysis of the two 6-h data sets ([11C]DHROL and [14C]IAP) demonstrated no relationship between the asymmetries of uptake of the two radiotracers (r2 = 0.099; p = 0.49). There were no significant differences observed in striatal weights (right vs. left), and the cerebellar [11C]DHROL uptake in MPP+-injected animals was equivalent to that seen in the saline-injected group (data not shown).
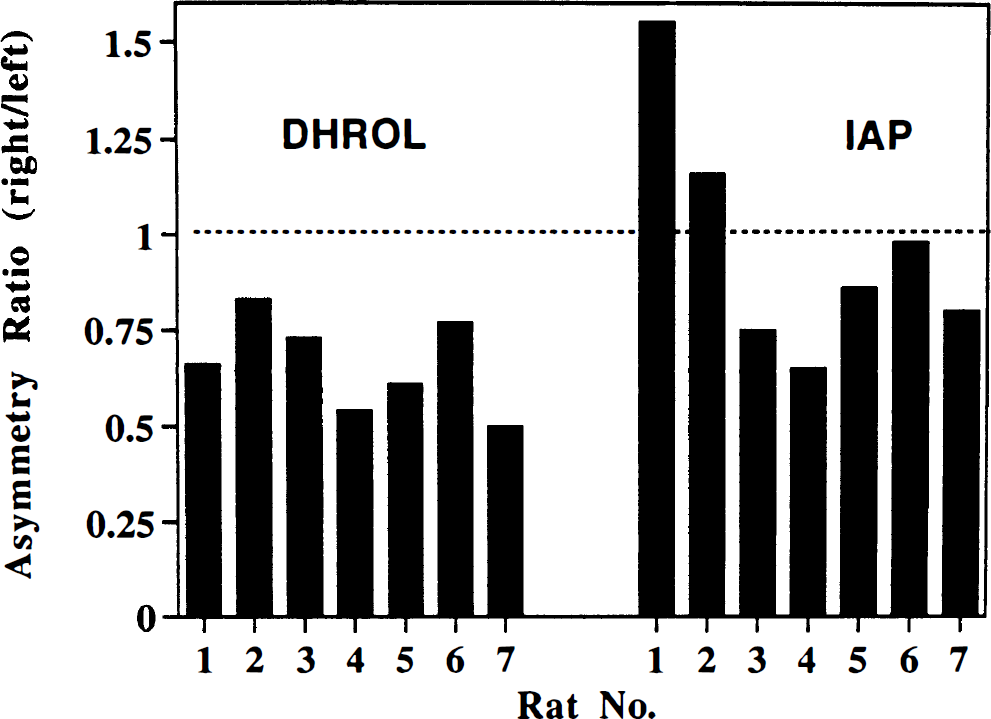
Asymmetry of striatal uptake of [11C]dihydrorotenol ([11C]DHROL) and [14C]iodoantipyrine ([14C]IAP) in rat brain 6 h after unilateral striatal injection of 100 nmoles MPP+. Data are shown as asymmetry ratios (% injected dose/g tissue, right/left striatum) for both radiotracers in each animal. Average asymmetry ratios were [11C]DHROL, 0.66 ± 0.05; and [14C]IAP, 0.98 ± 0.11. The asymmetries in [11C]DHROL and [14C]IAP were not correlated (p = 0.49).
DISCUSSION
Rotenone is a well-characterized inhibitor of mitochondrial complex I (Horgan et al., 1968; Singer and Ramsay, 1994). [11C]DHROL was developed as an imaging agent for in vivo investigation of complex I based on the successful in vitro autoradiographic imaging of this enzyme binding site in rat brain using [3H]DHR (Greenamyre et al., 1992). In vitro, DHROL competes for [3H]DHR binding to complex I with a fivefold lower potency compared with the parent compound, rotenone. The affinity of DHROL is similar to that of DHR, which has a Kd of about 90 nM (Higgins and Greenamyre, unpublished data). In vivo, [11C]DHROL shows a regional brain distribution similar to that obtained with [11C]rotenone itself. In addition, brain radioactivity after [11C]-DHROL injection is due entirely to authentic radiotracer: In separate chromatographic analyses of rat blood and brain tissues, authentic radiotracer ([11C]-DHROL) and polar metabolites were present in the blood, but only [11C]DHROL was found in the extracts of the brain tissue (data not shown). Thus, no metabolites of [11C]DHROL contribute to the radioactivity of brain tissues after i.v. injection of the radiotracer. Finally, the very high specific activities that can be achieved routinely with carbon-11 radiopharmaceuticals (> 1,000 Ci/mmol) should allow application of such labeled rotenoids in human studies: Rotenone is widely used as a pesticide and not particularly toxic (median lethal dose [LD50] i.v., rats, 0.2 mg/kg) and can be safely administered in the trace amounts (∼10 μg total) used in PET studies.
In the QA lesioned rat brain, there was a significant decrease (−40%) of in vivo [11C]DHROL retention in the injected striatum. In a subset of these animals studied using in vitro autoradiography, we found a similar (53%) loss of [3H]DHR binding. No changes of in vivo [11C]DHROL binding or in vitro [3H]DHR binding were evident in the animals injected with vehicle alone. The reductions of in vivo [11C]DHROL retention in the injected striata were significantly larger (>twofold) than those in CBF measured using [14C]IAP, even with the exclusion of the single animal with an unexpectedly higher blood flow in the injected side, suggesting that the losses of [11C]DHROL cannot be ascribed simply to decreases in radiotracer delivery to the lesioned striata.
MPP+ is a low-potency inhibitor of complex I and competes for [3H]DHR binding with an IC50 of about 5 mM (Greenamyre et al., 1992; Blandini and Greenamyre, 1995a). Intrastriatal administration of MPP+ also reduced in vivo [11C]DHROL uptake and retention, consistent with competition of MPP+ for the rotenone binding site on mitochondrial complex I. The reductions in radiotracer retention at 1.5 h after MPP+ infusion were variable and nonsignificant; however, at the 6-h time point, the losses of [11C]-DHROL retention became larger and more consistent, suggesting a requirement for sufficient diffusion of the neurotoxin throughout the striatum or across the mitochondrial membrane for full effect. As in the QA-lesioned animals, these changes in [11C]DHROL binding do not simply reflect decreases in tracer delivery, as blood flow was uniformly increased early after MPP+ injection and was on average unchanged at the later time point. In other words, changes in [11C]DHROL retention did not correlate with changes in blood flow.
The data from both the QA and MPP+ experiments are thus consistent with the notion that our new radioligand, [11C]DHROL, binds in vivo to the rotenone binding site of mitochondrial complex I. Use of intrastriatal MPP+ injection provides a model for direct, local inhibition of [11C]DHROL binding. Because mitochondrial enzymes are enriched in neurons versus glia, QA lesions provide a complementary model of neuronal degeneration, with accompanying loss of mitochondrial enzyme binding sites. Both acute MPP+ exposure and chronic quinolinate lesions produce a significant reduction of in vivo ([11C]DHROL) and/or in vitro ([3H]DHR) binding sites for radiolabeled rotenoids. For both MPP+ and quinolinate, the decreases in rotenoid binding are not due to the surgical trauma and are not simply a result of decreased blood flow to the injected striatum. Furthermore, our results are consistent with prior studies of intrastriatal neurotoxin injections. After MPP+ injections, measures of ATP production (reflecting intact oxidative phosphorylation) show a 50% decrease within 3 h, which increase to approximately 75% at 48 h (Storey et al., 1992). The trend toward the progressive reduction of [11C]DHROL binding at the later time point would be consistent with the slow accumulation of MPP+ in striatal neurons and then uptake into mitochondria (Ramsay et al., 1986b), with subsequent inhibition of ATP synthesis. Intrastriatal QA injections have been reported to cause a severe loss of neurons locally at the site of injection and a more widespread (∼50%) loss of cells in an area surrounding the central injection site (Roberts and DiFiglia, 1989). The losses of in vivo (−40%) and in vitro (−53%) rotenoid binding observed in our experiments are thus similar to these reported losses of cellular elements and ATP production.
Measuements of radiolabeled rotenoid binding should provide a new and different measure of metabolism in the diseased brain. Such measurements will be complementary to current PET studies of CBF, oxygen consumption or glucose metabolism (Snow, 1994), and 1H-nuclear magnetic resonance [1H-NMR] measurements of lactate levels (Jenkins et al., 1993). In preliminary experiments, we observed that the loss of in vivo [11C]rotenoid binding following QA lesioning was significantly larger (>twofold: p < 0.005, paired t test, n = 6) than the reduction of glucose metabolism measured using simultaneous in vivo injections of 2-[18F]fluoro-2-deoxy-D-glucose (Charalambous et al., unpublished data). A mismatch between glucose metabolism and oxidative phosphorylation might indicate a loss of ATP-generating capability by surviving cells, part of the proposed models of energy-dependent neurodegeneration.
The similar in vivo and in vitro reductions in rotenoid binding in the QA-lesioned animals suggest that [11C]DHROL, despite being a lipophilic molecule, does not suffer from a level of nonspecific binding which will obscure changes in specific binding in vivo. It is difficult to establish the extent of blockade of complex I through MPP+ injections, and the results of Storey et al. (1992) suggest that even with the injection of 100 nmol of MPP+ directly into the striatum, there is incomplete inhibition of complex I in the tissue (only 50% loss of ATP). Complete inhibition of the enzyme, even in a defined region through stereotactic injections, may be hard to accomplish and furthermore may be hard to separate from cell death. Thus, the residual [11C]DHROL uptake in MPP+-injected striata may reflect a contribution of nonspecific ligand binding; alternatively, it may indicate failure to block completely the rotenoid binding site. It is worth noting that studies of [3H]DHR binding suggest that nonspecific binding of this very similar ligand is low (∼20% of total) in preparation of intact platelets (Blandini and Greenamyre, 1995a, b ). In addition, we also determined that the radiotracer used in these experiments was a racemic mixture (isomers at the 5′-position): Recent studies have reported a 10-fold difference in in vitro binding affinities for the two 5′-isomers of rotenone (Ueno et al., 1994). Resolution of the inactive and active isomer of DHROL should provide a radiotracer with even lower nonspecific binding.
The changes in [11C]DHROL uptake in these experiments have been attributed to decreases in the numbers of available complex I binding sites due to neuronal loss (QA experiment) or occupation of the binding site (MPP+ injection). In the QA-lesioned animal, it is possible that the losses of [11C]DHROL retention are due to binding of the radiotracer to sites unrelated to complex I; however, no other high-affinity binding sites for rotenone have been reported in the literature, and our previous in vitro studies have shown a single binding site for radiolabeled rotenoids with a pharmacology and biochemistry. consistent with binding to complex I (Greenamyre et al., 1992). The losses of [11C]DHROL retention at the later time point after MPP+ infusion also could represent a contribution from cell death rather than occupation of the binding site by the neurotoxin. In an attempt to delay such cell death, we maintained the animals under ketamine anesthesia throughout the study in the hopes of preventing excitotoxicity; our current experimental design does not, however, allow us to ascribe definitively losses of [11C]DHROL only to blocking by the neurotoxin. Finally, a decrease in radiotracer accumulation also could result from a decrease in the in vivo binding affinity (Kd) of the radioligand, and from a single radiotracer injection, we cannot distinguish between these two possibilities. Age-related changes in the in vitro Kd for [3H]DHR in the mouse brain were reported recently (Higgins and Greenamyre, 1995), but no previous investigations into alterations of in vivo affinities for rotenoid binding to complex I in either aging or disease have been reported. The availability of an in vivo radioligand, such as [11C]DHROL, thus may allow the future evaluation of this possibility.
These studies support further investigation of radiolabeled rotenoids as in vivo imaging agents for studying complex I in the brain. Findings of altered postmortem complex I activity in human brains as a result of neurological disease (Beal et al., 1993; Bowling et al., 1994; Browne et al., 1994; Mann et al., 1994; Schapira and DiMauro, 1994; Beal, 1995), complex I inhibition as the mechanism of neurotoxicity of MPP+ (Mizuno et al., 1991; Nicklas et al., 1985), and possible (but controversial) detrimental effects of chronic levodopa (Przedborski et al., 1993, 1995; Cooper et al., 1995) or neuroleptic therapy (Burkhardt et al., 1993) on mitochondrial complex I activity support an interest in the in vivo evaluation of this enzyme complex in the living human brain.
In conclusion, we evaluated [11C]DHROL as a first-generation radioligand for the study of complex I in the rat brain. The striatal retention of this radiotracer was sensitive to prior occupation of the enzyme-binding sites by the competitive inhibitor, MPP+, or loss of cells through QA lesioning. Radiolabeled rotenoid inhibitors of complex I may thus provide important new tools for examining mitochondrial dysfunction in the human brain and the role of complex I in neurodegenerative diseases.