
Abstract
The muscarinic receptor antagonist scopolamine produces a transient memory deficit in healthy humans. This deficit has been offered as a model of the cholinergic deficit of Alzheimer's disease (AD). However, we have previously shown that scopolamine produces a deficit of cortical perfusion maximal in the frontal lobe, dissimilar to the parietal cortex deficit characteristic of AD. The current experiment was aimed at replicating and extending this observation by critically testing the central cholinergic origin of both cognitive and perfusion deficits. Nine healthy subjects participated in regional cerebral blood flow (rCBF) measurements at baseline, after scopolamine (7.2 μg/kg i.v.), and after both physostigmine (22 μg/kg i.v.) and neostigmine (7 or 11 μg/kg i.v.). rCBF was quantified by the xenon 133 inhalation method. As expected, scopolamine reduced cortical perfusion, mainly in the frontal cortex, and produced a memory deficit. Physostigmine, but not neostigmine, reversed all three variables partially or completely. These results support the hypothesis that all three consequences of scopolamine, namely, reduction of mean flow, frontal deficit, and memory impairment, are cholinergically mediated. Furthermore, because neostigmine poorly crosses the blood–brain barrier, these findings confirm that the effect is centrally mediated and cannot be explained by peripheral effects. However, they also confirm the frontal cortex locus of action for both scopolamine and its reversal by physostigmine and therefore suggest a major dissimilarity to the characteristic rCBF appearance of AD. This study extends our previous preliminary findings with tacrine and strengthens the suggestion that only nicotinic receptors are associated with the characteristic parietal deficit of AD.
Keywords
Scopolamine, a muscarinic receptor antagonist, induces a transient memory deficit in healthy humans (Drachman and Leavitt, 1974). This effect has been proposed to mimic Alzheimer's disease (AD) in normals, and the different responsivity of AD patients is suggested to support a cholinergic contribution to its pathophysiology (Molchan et al., 1992; Aarsland et al., 1994; Neufeld et al., 1994). It is especially critical to validate the “scopolamine model” of AD, since it is now being increasingly proffered for purposes of drug screening and development (Wesnes et al., 1991; Christensen et al., 1992; Sahakian et al., 1993). We previously reported a problem in this model. AD is characterized by a typical parietotemporal deficit in cortical metabolism and blood flow (Friedland et al., 1983; Jagust et al., 1987; Prohovnik et al., 1988). In contrast, we (Honer et al., 1988) have shown that scopolamine causes a frontal, rather than a parietotemporal, cortical perfusion deficit. Therefore, scopolamine-induced amnesia appears to depend on cerebral networks different from those affected by AD. This suggestion is consistent with reported discrepancies between the neuropsychological profile of AD and that of scopolamine-induced deficits in normals (Lines et al., 1991) and differences between scopolamine effects on visual evoked responses and the effects of AD (Ray et al., 1991).
We previously replicated the hypofrontal scopolamine effect on cerebral perfusion in a pharmacologically complex situation, following nicotinic blockade (Gitelman and Prohovnik, 1992). The current experiment was undertaken to directly replicate the scopolamine effect and verify its central cholinergic mechanisms. Specifically, we tested whether the amnesic and regional cerebral blood flow (rCBF) effects of scopolamine could be attributed to nonspecific side effects or systemic factors, rather than a specific CNS anticholinergic effect. The current design replicates our previous experiments but uses physostigmine (a centrally active cholinesterase inhibitor) and neostigmine (a peripheral cholinesterase inhibitor) for reversal following scopolamine injections. Since physostigmine has demonstrated efficacy in the treatment of AD and has been claimed to reverse the AD-like cognitive deficits induced by scopolamine (Wesnes et al., 1991), we tested whether such a reversal would occur in our model and whether it would be accompanied by reversal of the rCBF abnormality. The neostigmine condition provided a control for the peripheral cholinergic agonism of physostigmine.
METHODS
Other than changes related to the pharmacological reversals, all procedures were identical to those employed by Honer et al. (1988). All time points are denoted in relation to scopolamine injection, unless otherwise noted.
Subjects
Ten healthy young subjects were recruited. One was dropped owing to marked hypotensive bradycardia in response to neostigmine, yielding a final sample size of nine. All were male, with a mean age of 25 ± 5 years (range 19–35 years).
Design
All subjects visited the laboratory for initial screening and then twice for the experimental procedures. The procedures were identical on both days and were performed at the same time of day, except for the randomized order of either physostigmine or neostigmine. Baseline procedures included the Buschke Selective Reminding Test (BSRT; Buschke, 1973) and rCBF measurement by the Xenon 133 inhalation technique. Scopolamine (7.2 μg/kg i.v.) was then administered, and a second rCBF (run 2) was begun 5 min after the injection. Two minutes before the end of this procedure, a slow infusion of either physostigmine (22 μg/kg) or neostigmine was started, which lasted 10 min. The first five subjects received a neostigmine dose of 11 μg/kg i.v.; the other four were given 7 μg/kg. All subjects received both neostigmine and physostigmine on separate days in randomized order. The third rCBF procedure (run 3) started at the end of the infusion, 25 min after scopolamine injection. Then the BSRT was again administered, and the final rCBF determination was made 75 min from scopolamine injection. The BSRT was repeated at ∼90 min after scopolamine injection.
The main point of interest is run 3, the third rCBF measurement. Occurring at ∼25 min from scopolamine administration and at the end of the reversal infusion, it should reflect the peak additive effects of scopolamine plus physostigmine versus scopolamine plus neostigmine, compared with run 2. Figures 1 and 2 give a graphic depiction of the experimental design.
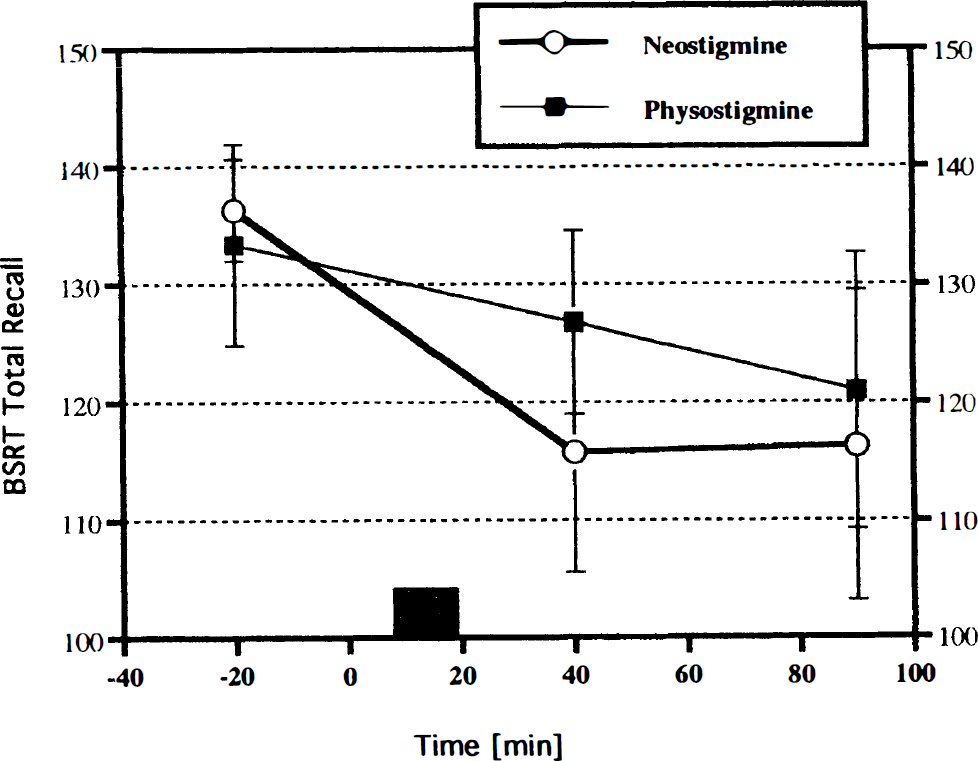
Buschke Selective Reminding scores (total recall, mean ± SD) by time (minutes relative to scopolamine injection) and AChE inhibitor (physostigmine versus neostigmine). The striped rectangle indicates the time of AChE inhibitor infusion. Scopolamine impairs recall, but the impairment is briefly ameliorated by physostigmine infusion.
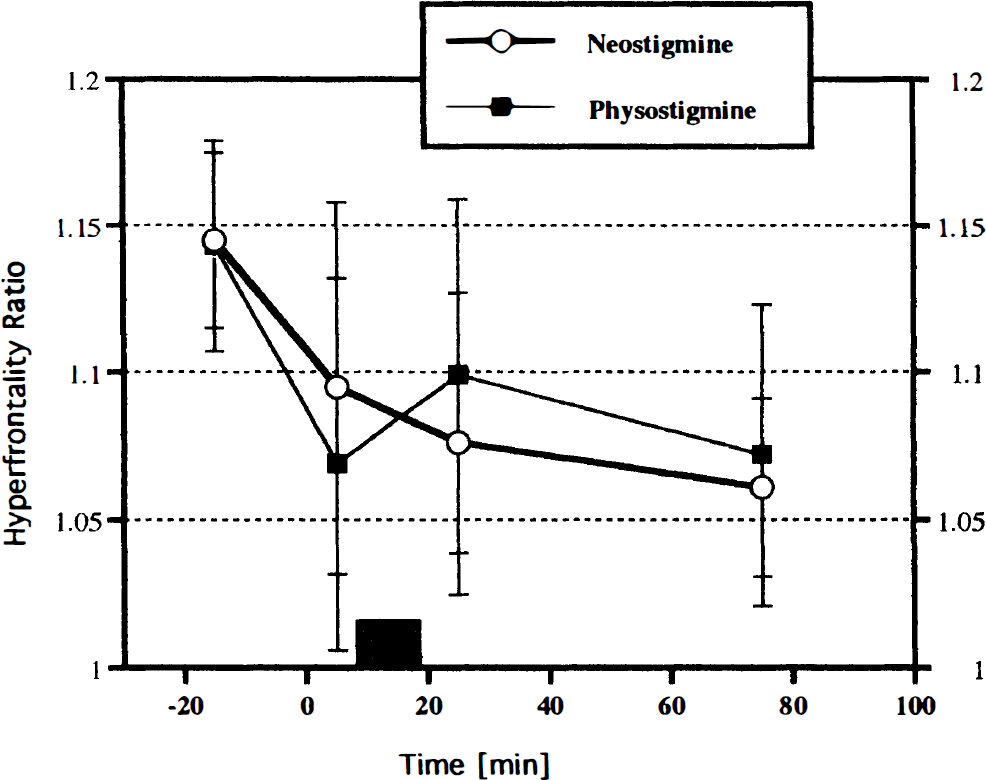
Hyperfrontality ratio (frontal cortex divided by posterior cortex, mean ± SD) by time (minutes relative to scopolamine injection) and AChE inhibitor (physostigmine versus neostigmine). The striped rectangle indicates the time of AChE inhibitor infusion. Scopolamine reduces hyperfrontality, but the reduction is briefly ameliorated by physostigmine infusion.
rCBF
Procedures were identical to those described by Honer et al. (1988). Each cerebral hemisphere was covered by 16 detectors, and pulse rate, blood pressure, and end-tidal P
Data analysis
As in our previous experiments, significant main effects or interactions involving the two hemispheres were not detected, so all regions were bilaterally averaged. The two dependent rCBF variables of central interest for quantitative analysis were the global mean flow and the hyperfrontality (anterior/posterior) ratio. Anterior locations were the regions denoted F1, F2, F3, F4, and F5 in the standard helmet, located over the frontal lobes, whereas posterior locations included all other 11 regions (from a total of 16 cortical regions). This division was examined because it had emerged as the major effect of scopolamine in the previous experiment, and a topographic visualization can be found in the previous report (Honer et al., 1988).
Data were analyzed by repeated-measures and multivariate analyses of variance (ANOVA, MANOVA) techniques (for which the conservative Greenhouse–Geiger corrected p is provided), with the neostigmine dose (high/low) as a between-subject factor and drug (physostigmine/neostigmine) and run (1–4) as repeated measures. Results are reported with the F ratio, degrees of freedom, and significance level. Specific contrasts are reported as conservative unplanned comparisons, rather than planned post hoc analyses, to further reduce type I error. Analyses were performed with the SuperANOVA package on a Macintosh computer. BSRT scores reflect total immediate recall. Numerical data are provided in the text as the mean ± SD.
RESULTS
Cardiovascular and respiratory changes are provided in Table 1. Systolic blood pressure rose slightly after scopolamine and decreased following cholinesterase inhibition, but neither the main time effect nor any interactions approached significance. Diastolic blood pressure showed similar nonsignificant trends. P
Respiratory and cardiovascular measures (mean ± SD) during the eight conditions
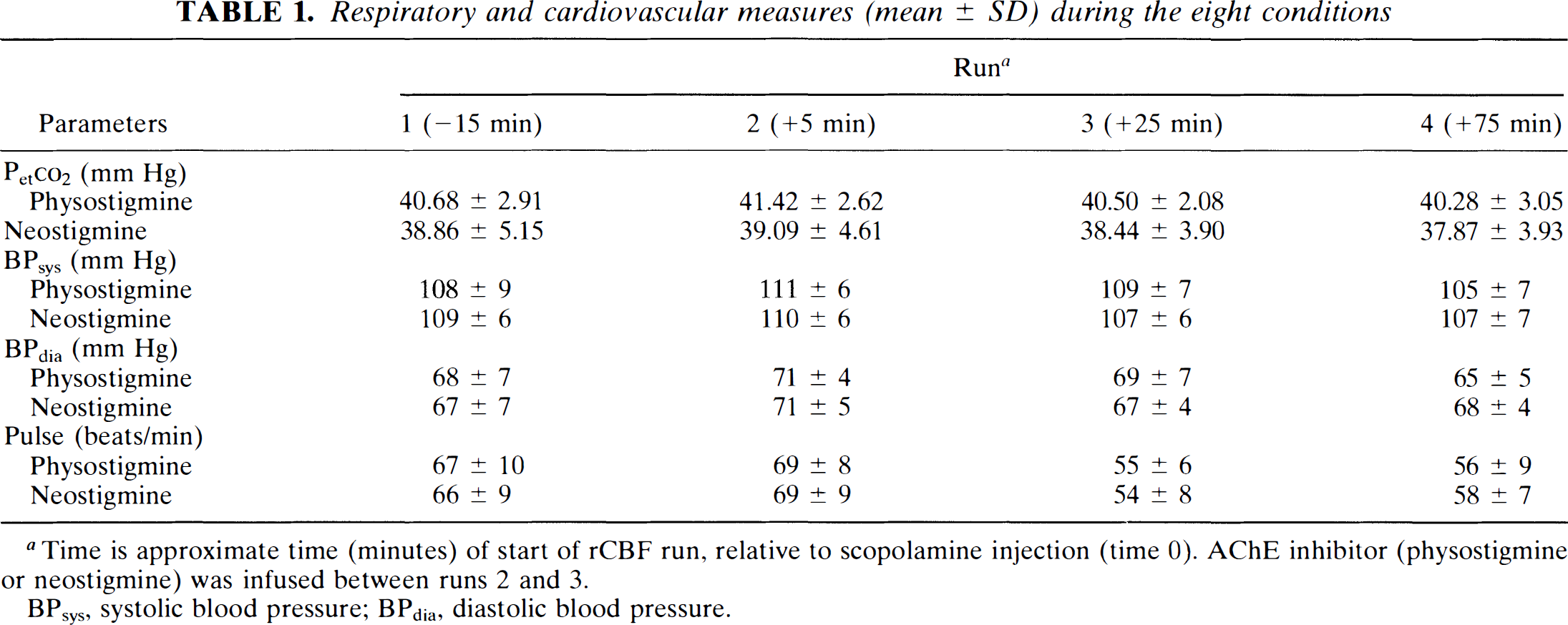
Time is approximate time (minutes) of start of rCBF run, relative to scopolamine injection (time 0). AChE inhibitor (physostigmine or neostigmine) was infused between runs 2 and 3.
BPsys, systolic blood pressure; BPdia, diastolic blood pressure.
BSRT total recall, the measure of cognitive drug effect, showed a significant time effect (F2,16 = 20.00, p = 0.0003) and a significant time by drug interaction (F2,16 = 4.06, p < 0.05). To further examine the interaction, a separate analysis was performed on the second BSRT session at 40 min. There was a significant difference between physostigmine and neostigmine (F1,7 = 26.22, p = 0.001), with physostigmine yielding better BSRT performance than neostigmine (127 vs. 116). Of note, there was also a significant interaction with neostigmine dose (F1,7 = 13.27, p < 0.001): BSRT performance on the high neostigmine dose was almost as good (124) as with physostigmine, whereas it was substantially lower on the low neostigmine dose (109). However, this was a specific effect for this time point, and there was no main effect or interaction with neostigmine dose in the main analysis. The data are summarized in Fig. 1.
For mean cortical perfusion, the only near-significant effect was a drug by time interaction (F3,24 = 3.69, p < 0.06 with Greenhouse–Geiger and <0.03 without). At the critical comparison of run 3 to run 2, after physostigmine, flows were increased (from 64.16 ± 9.36 to 69.12 ± 7.20), whereas they decreased following neostigmine (from 73.21 ± 12.60 to 67.79 ± 7.72). This difference was robust (F1,7 = 10.28, p = 0.01). In comparison with the baseline condition as well, flow was higher following physostigmine and lower following neostigmine, but this comparison was nonsignificant.
The hyperfrontality ratio (Fig. 2) showed a significant change with time (F3,24 = 11.70, p < 0.001) and a drug by time interaction (F3,24 = 3.59, p < 0.05). There was an immediate and marked reduction in this ratio following scopolamine. However, there was a short-lived increase of this ratio after physostigmine (from 1.076 to 1.104); neostigmine was associated with a continuous decline at this point (from 1.097 to 1.077). This difference was significant (F1,7 = 7.43, p < 0.03). Specific post hoc contrasts between the drug conditions showed a significant difference only during the third, post-physostigmine run (F1 = 6.21, p < 0.03); the three other conditions were not significantly different. The complete regional change over the cortex from run 2 to run 3 is depicted in Fig. 3.
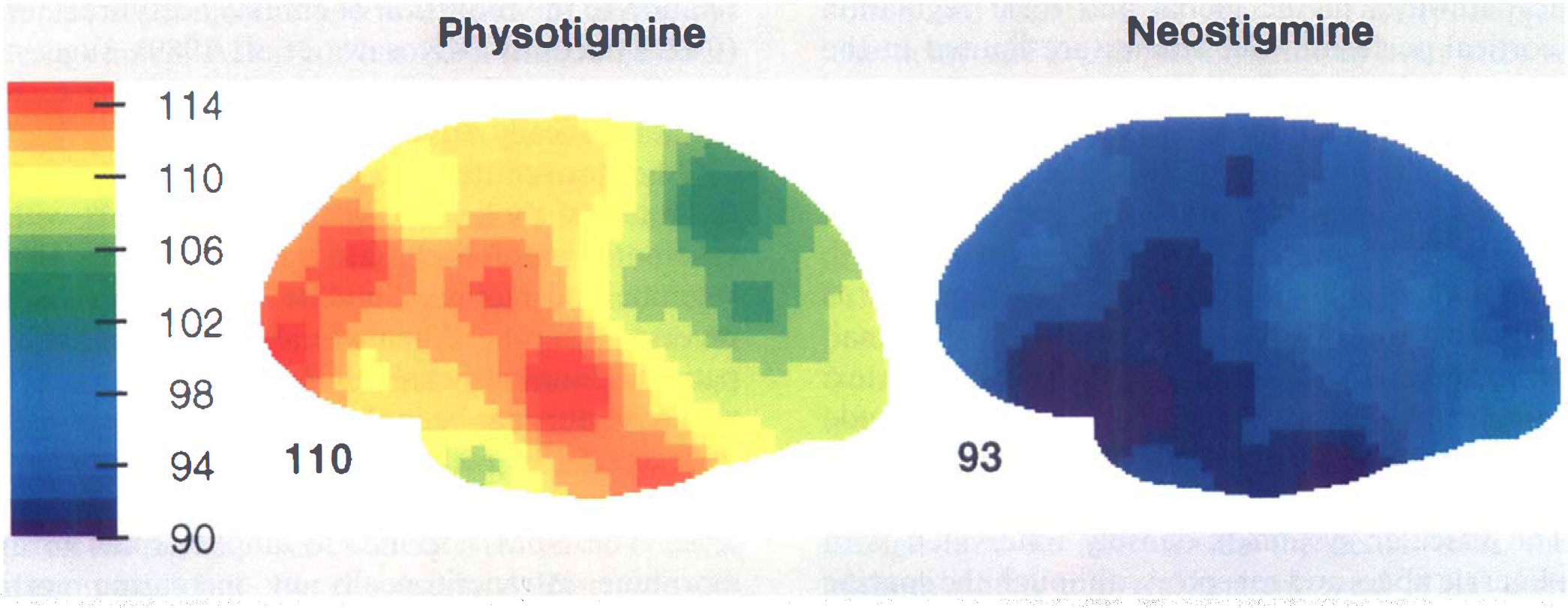
Changes of cortical blood flow (ml/100 g/min) caused by a 10-min infusion of neostigmine or physostigmine, 5 min after injection of scopolamine (7.3 μg/kg). Data are group means for all subjects and conditions (including both neostigmine doses), depicted as the percentage change of regional flow from before to after AChE inhibitor infusion (100 equals no change). The numbers below depictions denote mean brain changes of flow (+ 10% for physostigmine, −7% for neostigmine). Note that physostigmine, but not neostigmine, increases hyperfrontality.
DISCUSSION
Replicating our previous reports (Honer et al., 1988; Gitelman and Prohovnik, 1992), we found scopolamine reduced global cortical perfusion and hyperfrontality as well as immediate selective recall. Physostigmine, but not neostigmine, briefly reversed all three changes. The memory deficit induced by scopolamine is a ubiquitous finding (Wesnes et al., 1991; Blin et al., 1994). We have previously suggested that this deficit is associated with frontal cortex dysfunction, based on concurrent rCBF measurements in human subjects (Honer et al., 1988). The reversal of the memory deficit and frontal cortex hypoperfusion by physostigmine adds strong support to this hypothesis. The following discussion will concentrate on the novel observations of neostigmine, limitations of the current study, remaining discrepancies with previous reports, and the mechanism of action of physostigmine.
Neostigmine effects
We were surprised to find the apparent central effects of neostigmine at the higher dose. This medication was selected for its poor penetration of the blood–brain barrier. Like other quaternary ammonium compounds, neostigmine is strongly believed to be active only peripherally (c.f., Goodman Gilman et al., 1985; Standaert, 1990) and therefore should provide an effective control for the peripheral scopolamine-induced side effects. Of course, massive doses could overcome the poor penetration, but the dose we used is highly conservative and below what was used in other clinical and research reports. Furthermore, the identical autonomic effects of the two cholinomimetics in this study confirm that the neostigmine dose was correctly chosen to be peripherally equipotent to physostigmine. Indeed, in its routine use to reverse neuromuscular blockade in anesthesia, neostigmine is administered, to maximally inhibit AChE, as an intravenous bolus at doses of 35–70 μg/kg, or three to 10 times higher than the doses we used as a slow infusion. Despite these relatively high doses, anesthesiologists believe the drug is not centrally active. This suggestion is supported by behavioral studies in awake psychiatric patients to evaluate the cholinergic theory of depression; neostigmine has been compared with physostigmine and showed no effects (Schaffer et al., 1981). Janowsky et al. (1973a) compared 1.25 mg neostigmine (i.v. over 25 min) with 2.50 mg physostigmine (i.v. over 25 min). For a standard 70-kg adult, these doses correspond to ∼18 and 36 μg/kg, >50% higher than our doses. Nevertheless, these authors found no behavioral effects of neostigmine either by itself or as an antagonist for methylphenidate, whereas physostigmine was effective in both.
In another article these authors (Janowsky et al., 1973b) report administering ≤1.5 mg of neostigmine over 15 min, corresponding to ∼21 μg/kg, to manic patients. Bradycardia was observed, but not nausea or vomiting, and there was no significant change in behavioral rating scales, although in some patients the authors noted a “minimal transient sluggishness.” Thus, it is difficult to reconcile our apparent central effects with their absence in previous literature. Several factors are known to modulate neostigmine potency, including acid-base balance (especially if due to respiratory acidosis) and electrolyte balance: no such confounders are expected in our young healthy subjects. Had we observed only vascular effects, we could have hypothesized vasomotor action through receptors external to the blood–brain barrier, but the amelioration of memory deficit, if true, requires intracerebral action. Therefore, this is a type 1 error due to the small sample size, or else neostigmine penetrates the barrier more efficiently than previously suspected. Further research is in progress to evaluate this possibility.
Limitations of the current study
Although these and previous findings directly implicate the cholinergic system, and specifically muscarinic pathways, in the global and focal regulation of cortical perfusion, our studies are limited in the separation of metabolic versus vascular effects and cannot exclude an intervening or interacting noncholinergic mechanism. In addition, the current study cannot be generalized to older subjects; as we (Gitelman and Prohovnik, 1992) and others have noted, aging modifies cholinergic responsivity. Finally, it is not possible to exclude the possibility of very small physostigmine effects in the parietotemporal cortex: although these effects must be of lesser magnitude and smaller spatial extent, they may be detectable by higher-resolution methods.
The vascular system is densely innervated with cholinergic fibers and receptors, although the specific receptor subtypes are still being debated (Kalaria et al., 1994). Cholinergic stimulation or antagonism can change cortical perfusion by affecting metabolism and secondarily driving blood flow. Alternatively, cholinergic manipulation may alter the coupling mechanisms between metabolism and blood flow or directly change vascular resistance. Cortical microvessels are innervated by cholinergic fibers that do not originate in the nucleus basalis of Meynert (NbM) but in intrinsic cortical cholinergic neurons (Galea et al., 1991). It has been shown in rabbits that carbachol (a cholinergic agonist) increases caudate nucleus perfusion by direct action on muscarinic receptors of vascular smooth muscle as well as inhibition of sympathetic, noncholinergic, constrictory tone (Aubineau and Sercombe, 1977). On the other hand, cholinergic pathways are known to be involved in the cortically intrinsic neurogenic control of blood flow by metabolism and to be affected by cholinergic agents (Scremin et al., 1973). The current study cannot determine whether the vascular changes observed are entirely or in part metabolically driven, but in rats, physostigmine may be able to enhance perfusion without any changes of metabolism (Scremin et al., 1990).
A common model to test cholinergic mechanisms consists of stimulation of ablation of the NbM or its rodent equivalent, the substantia innominata, both providing the main source of cortical cholinergic input. Vaucher et al. (1995) have recently suggested that NbM stimulation in the conscious rat causes a frontal cortex flow enhancement that is much stronger than the glucose metabolic increase, whereas in parietal cortex they found weaker flow enhancement without metabolic increases. The cortical metabolic effects of NbM stimulation are yet to be resolved (Sato and Sato, 1992; Fukuyama et al., 1995). In contrast, lesions of the NbM are well known to cause reductions of cortical glucose utilization, predominantly in the frontal cortex and in direct correlation to the reduction of choline acetyltransferase (ChAT) activity (Kiyosawa et al., 1989), suggesting that the physostigmine effects observed here could be metabolically driven.
Other transmitter systems are well known to affect the cerebral cholinergic system and interact with its vasomotor regulation (Sato and Sato, 1992). Physostigmine and methylphenidate have been shown to reverse each others' behavioral effects in psychiatric patients (Janowsky et al., 1973a). A scopolamine dose similar to ours has been shown by Dewey et al. (1993) to decrease [11C]raclopride binding in the human striatum, consistent with an increase in dopamine release. The NbM responds to amphetamine or apomorphine intraperitoneally by increasing cortical acetylcholine (ACh) output (Sato and Sato, 1992). Thus, we cannot yet exclude the possibility that some of our observations are due to indirect effects of other neurotransmitter systems, although they are also consistent with pure cholinergic mechanisms. In particular, as mentioned earlier, there is yet no established anatomical substrate to the apparent frontal predominance of scopolamine and physostigmine effects; this finding may be due to other interactions (e.g., dopaminergic).
Comparison with previous reports
There is a dearth of literature concerning these effects in humans, and the only previous article that addressed both scopolamine and physostigmine yielded unexpected findings. Because it is the only such experiment in the literature, it is reviewed in some detail. Using 2-deoxy-2-fluoro-
The current study confirms that physostigmine's effects are rapid and transient. The behavioral protective effect was noticeably weaker by 90 min (Fig. 1). The effects on mean cortical perfusion and hyperfrontality were evident only during run 3 and completely dissipated 50 min later (Fig. 2). Dauphin et al. (1991) reported that cortical perfusion increases induced by physostigmine (in rats) disappeared within 30 min of the end of infusion. Similarly, Molnar et al. (1991) found in rabbits that physostigmine infusion (60 μg/kg i.v. over 10 min) increased cortical perfusion by ∼50%, but the maximal effect was observed at 17 ± 2 min, and CBF returned to baseline by 32 ± 4 min from the end of infusion. Scremin et al. (1990) showed (in rats) by iodoantipyrene autoradiography similarly fast changes in response to 50 μg/kg of physostigmine: perfusion was elevated by 5 min and began to return to baseline by 12 min. In fact, these authors showed that the elimination half-life of physostigmine from most cortical areas (other than occipital and olfactory) ranged from 4 to 10 min. The FDG procedure is usually insensitive to rapid changes, and Blin et al. (1994) were forced to reduce the dose and prolong the scan further to allow for two measurements. Therefore, a transient effect could have been missed. Finally, the doses of physostigmine were dissimilar: whereas we infused 22 μg/kg over 10 min, Blin et al. infused only 5 μg/kg/h. We agree with their caution in using physostigmine in AD patients, since we have run into similar problems (Wirkowski et al., 1991). However, it is likely that these methodological problems all limit the power of their very weak physostigmine findings.
Cholinergic muscarinic blockade in general, and scopolamine in particular, are virtually universally stated to reduce cerebral metabolism and blood flow in textbooks (e.g., see references in McCulloch, 1988; Edvinsson et al., 1993; Kurumaji et al., 1993; but see also exceptions, such as Dauphin et al., 1991). It is, therefore, difficult to explain the paradoxical increase of cortical metabolism by scopolamine reported by Blin et al. (1994). Their dose of 0.25 mg/m2 of body surface area would result in about 0.43 mg in a standard adult, ∼20% lower than our dose of 7.2 μg/kg; furthermore, they administered it by slow infusion, whereas ours was a bolus injection. In our original experiment (Honer et al., 1988), we showed that the dose/response curve was rather steep at these ranges, and, at least in young subjects, this difference could be consequential. Ray et al. (1991) documented an equally strong difference between the P100 visual evoked potential effects of 4 and 7 μg/kg doses in young and middle-aged (but not elderly) subjects. The delay of FDG injection until an hour after scopolamine administration, and the low temporal resolution discussed earlier, would further reduce the effect.
Blin et al. (1994) acknowledge that their results are inconsistent with rCBF findings in humans and FDG findings in rodents and suggest, among other possibilities, that scopolamine decouples perfusion from glucose metabolism in humans. Such decoupling is also indicated by the recent report of Ogawa et al. (1994), who measured both CBF and CMRglc in cats with PET during somatosensory stimulation. Although they do not provide the actual data, these authors stated that the contralateral rCBF activation was abolished by scopolamine (350 μg/kg i.v.), whereas the metabolic response was preserved. However, Ogawa et al. (1994) argue only that scopolamine changes the activation response coupling, not the baseline tone: according to their results, scopolamine does reduce resting metabolism and blood flow. In humans, however, Molchan et al. (1994) also failed to find substantial reductions of glucose metabolism (by FDG–PET) after 0.5 mg of scopolamine (i.v.). That group did report weak reductions in prefrontal cortex, consistent with the location of our findings. Thus, although there are similarities in FDG–PET findings for both physostigmine and scopolamine, they are weaker and require further clarification.
Mechanisms of action
This is the third observation, and second independent replication, in the frontal locus of scopolamine-induced perfusion deficits in healthy humans. We have previously shown that this effect is dose-dependent (Honer et al., 1988) and can be seen in both young and elderly subjects (Gitelman and Prohovnik, 1992). The anatomical substrate for this topographic specificity is unknown and is unlikely to be based on denser cholinergic projections or receptor concentrations in frontal cortex: we could not find any evidence in the literature of frontally increased ChAT or ACh binding in normal human brains. However, there is evidence from animal experiments that the frontal cortex is most sensitive to stimulation of the substantia innominata (SI) (Dauphin et al., 1991; Vaucher et al., 1994). In conscious rats, SI lesions reduce ChAT activity mostly in the frontal cortex but do not reduce the flow to the same extent or in exactly the same regions (Peruzzi et al., 1993). Physostigmine, in the same experiment, caused widespread cortical vasodilatation independent of SI lesions. The authors concluded that physostigmine does not act through the SI system but, instead, directly on cortical interneurons. Alternatively, it was recently reported that the density of nitric oxide synthase–positive neurons in the frontal cortex of the rat is significantly higher than in other cortical regions; these neurons bear muscarinic receptors and may mediate the vascular effects projecting from the basal forebrain (Moro et al., 1995). Physostigmine distribution in the rodent brain has been studied (Scremin et al., 1990), and the flow elevations do not correspond to areas of high initial concentration, that is, they are not a function of accessibility and availability. The current findings of reversal of scopolamine-induced hypofrontality with physostigmine, but not neostigmine, offer strong support for the centrality of the frontal cortex in the responses of the human brain to cholinergic manipulations.
Our study also sheds new light on the cerebral action of physostigmine. Because this drug shows a therapeutic benefit in AD, authors previously attempted to find that it also ameliorated the parietotemporal perfusion deficit characteristic of that disease. Although Gustafson et al. (1987) could not document any absolute CBF changes after infusion of physostigmine (28 μg/kg over 2 h), they reported significant relative increases in the parietotemporal cortex. These increases, however, were not significant compared with baseline values (but only compared with placebo-induced decreases) and were obtained only by separate computation of this single location, without an omnibus analysis. Similarly, Geaney et al. (1990) reported relative increases of parietotemporal perfusion in AD patients, but not in controls, in response to intravenous infusion of ∼7 μg/kg over 30 min. In contrast, Hunter et al. (1991) suggested frontal, especially left frontal, flow enhancement with a similarly low dose.
Our study is the first to quantify a frontal cortex effect in tight association with a memory test effect. There is other preliminary evidence, though far from definitive, that the frontal cortex is a major target for cholinergic manipulations involving AChE inhibition. Ebmeier et al. (1992) reported a relative increase of perfusion (uptake with single photon emission computed tomography) in the superior frontal cortex and a decrease in the left parietotemporal cortex, in response to a dose of velnacrine, another AChE inhibitor, that slightly improved word recognition memory. We have shown such effects with tacrine, the only medication currently approved for treatment of AD. In a preliminary study (Prohovnik et al., 1994), we measured rCBF in three patients who received 80 mg tacrine orally for 8 weeks. Mean cortical perfusion rose by ∼10%. Preliminary FDG–PET data are consistent with this finding (Nordberg, 1993). The greatest and most consistent changes were seen in frontal cortex areas. Finally, there is increasing clinical and neuropsychological evidence that such drugs, and especially tacrine, exert their maximal effect on attentional mechanisms, rather than the core amnestic syndrome of AD (Sahakian et al., 1993); this finding would suggest a frontal cortex locus of action.
In conclusion, this experiment fully replicates the scopolamine-induced frontal deficit first reported by Honer et al. (1988). Moreover, this experiment confirms that both the perfusion hypofrontality and the memory deficit induced by scopolamine in young healthy subjects are reversible by physostigmine, but not neostigmine. Therefore, both depend on centrally mediated cholinergic effects, rather than peripheral side effects, and seem to be primarily related to the frontal cortex. We do not yet have an explanation for the frontal site of action, and it remains to be established whether these findings support or refute the “scopolamine model” of AD. However, to the extent that the scopolamine hypocholinergic state is a valid simulation, our findings suggest that the beneficial effects of physostigmine and, by extension, other AChE inhibitors depend on frontal lobe mechanisms and may specifically improve those higher cognitive functions, such as attentional mechanisms, that indirectly affect memory performance. In this area, our findings support recent observations on the therapeutic actions of tacrine in AD.
Footnotes
Acknowledgment:
Research was supported in part by a grant from the Jean and Louis Dreyfus Foundation and PHS grant AG05433.