
Abstract
In acute brain injury syndromes, the potent N-methyl-D-aspartate (NMDA) antagonist, MK-801, can prevent neuronal degeneration, and the general anesthetics, isoflurane and propofol, may also provide neuroprotective benefits. An obstacle to the use of NMDA antagonists for neuroprotective purposes is that they can cause a neurotoxic vacuole reaction in cerebrocortical neurons. This study demonstrates the ability of isoflurane and propofol to prevent the neurotoxic vacuole reaction induced by MK-801. Low sedative doses of inhaled isoflurane (1%) or intravenous (i.v.) propofol (7.5 mg/kg/h) were as effective as higher general anesthetic doses. Thus, in the clinical management of acute brain injury conditions such as stroke and brain trauma, administration of one of these anesthetic agents together with an NMDA antagonist may be an excellent formula for obtaining optimal neuroprotection while eliminating serious side effects.
Excessive activation of excitatory receptors by the endogenous transmitter, glutamate, is believed to trigger neuronal degeneration in commonly occurring acute brain or spinal cord injury syndromes (stroke, trauma, hypoglycemia, status epilepticus) (Choi, 1988). The N-methyl-
A potential obstacle to the use of NMDA antagonists for neurotherapeutic purposes is the fact that these agents cause psychotomimetic reactions in adult humans (Luby et al., 1962; Domino and Luby, 1981; Reich and Silvay, 1989; Krystal et al., 1993) and can injure or kill cerebrocortical neurons in the adult rat brain. At low doses, NMDA antagonists cause a reversible vacuole reaction in pyramidal neurons of the posterior cingulate and retrosplenial (PC/RS) cortex (Olney et al., 1989), but at higher doses, PC/RS neurons are killed (Fix et al., 1993; Corso et al., 1994). In order for NMDA antagonists to be of optimal value as neuroprotective agents, it will be necessary to develop strategies for counteracting or avoiding these side effects.
Ketamine, an NMDA antagonist that has been used in human anesthesia for two decades, causes psychotic (“emergence”) reactions in human adults that are reduced in frequency and severity by coadministration of either of two classes of drugs—benzodiazepines or barbiturates—that facilitate γ-aminobutyric acid (GABAA) receptor-mediated neurotransmission (Magbagbeola and Thomas, 1974; Reich and Silvay, 1989). It has been shown that these two classes of drugs also prevent the neurotoxic vacuole reaction induced in rat cerebrocortical neurons by the powerful NMDA antagonist, MK-801 (Olney et al., 1991). Based on this and other related evidence, it has been proposed that a GABAA receptor-mediated disinhibition mechanism underlies both the psychotic and neurotoxic side effects of NMDA antagonists (Olney and Farber, 1995). Further supporting this proposal is the recent finding by Ishimaru et al. (1995) that halothane, a volatile anesthetic that is thought to facilitate GABAA neurotransmission, prevents MK-801-induced neurotoxicity.
In the present study, two additional anesthetic agents, isoflurane and propofol, were evaluated for their ability to prevent MK-801 neurotoxicity. Isoflurane is an agent with anesthetic properties similar to those of halothane, but with fewer untoward side effects, especially in a neurosurgical setting (Stevens and Kingston, 1992). Propofol (2,6-diisopropylphenol) is a well-tolerated intravenously (i.v.) administered anesthetic, which has been used widely in human anesthesia in the past decade. Both isoflurane and propofol are thought to act by a mechanism involving GABAA receptors (Nakahiro et al., 1989; Hara et al., 1993).
MATERIALS AND METHODS
Adult female Sprague–Dawley rats (weight 290–320 g) were used for all experiments. MK-801 (dizocilpine) (Research Biochemicals Incorporated, Natrick, MA, U.S.A.) was the NMDA antagonist used in all experiments. The general design of experiments was to administer MK-801 in a subcutaneous (s.c.) dose (0.5 mg/kg) that consistently induces a specific pattern and degree of cerebrocortical damage and to determine whether administration of either isoflurane or propofol together with MK-801 would modify the neurotoxic reaction.
For the isoflurane experiments, both control and experimental rats received MK-801 (0.5 mg/kg, s.c); experimental rats were additionally exposed to isoflurane at various concentrations and according to several time schedules. Isoflurane vapor was administered through a nose cone at an air flow of 2 L/min. A newly calibrated isoflurane vaporizer (Ohio Medical Products, Madison, WI, U.S.A.) was utilized so that the dialed concentration closely corresponded to the percent of isoflurane vapor actually administered.
For the propofol experiments, both control and experimental rats received MK-801 (0.5 mg/kg, s.c); experimental rats additionally received propofol (Zeneca Pharmaceuticals, Wilmington, DE, U.S.A.) i.v. through a tail vein. At 15 min before administration of MK-801, propofol was given in an i.v. bolus dose (20 mg/kg), and this was followed by a steady i.v. infusion of propofol at one of several doses and according to one of several time schedules. The i.v. infusion was delivered via a 24-guage angiocath connected to a syringe pump (Sage Model 352 pump, Orion Research Incorporated, Boston, MA, U.S.A.) that administered the desired infusion rate (calculated as mg/kg/h).
The duration of treatment in the above experiments varied from 4 to 12 h. At the termination of the experiment, the rats were deeply anesthetized with pentobarbital and killed by perfusion through the left cardiac ventricle with a phosphate-buffered fixative solution containing 4% paraformaldehyde and 1.5% glutaraldehyde. After the perfusion, the brains were removed from the skull and cut transversely into 1-mm thick slices. The slices were post-fixed in 1% osmium tetroxide, dehydrated in ethanol, cleared in toluene, and embedded in araldite. Sections were cut 1 μm thick using 0.5-in wide glass knives on a Sorvall MT-2B 0.5-in ultratome. Sections were stained for light microscopic evaluation with a mixture of methylene blue and azure II. The severity of the neurotoxic reaction was assessed by counting the number of vacuolated neurons present in sections cut through the PC/RS cortex at a specific rostrocaudal level (6 mm caudal to bregma), where the toxic reaction is known to be maximally expressed.
RESULTS
In the isoflurane experiments, we found that prevention of vacuole formation depends on the relative timing of the isoflurane and MK-801 treatments. When isoflurane was administered for 1 h (1.5–2.0% inhaled concentration) and then discontinued immediately prior to MK-801 treatment, the number of vacuolated neurons in the PC/RS cortex 4 h after MK-801 was similar to that in animals treated with MK-801 alone. In contrast, if isoflurane was administered 15 min prior to MK-801 and continuously thereafter until death 4 h later, there were no vacuolated neurons in the PC/RS cortex (Fig. 1).
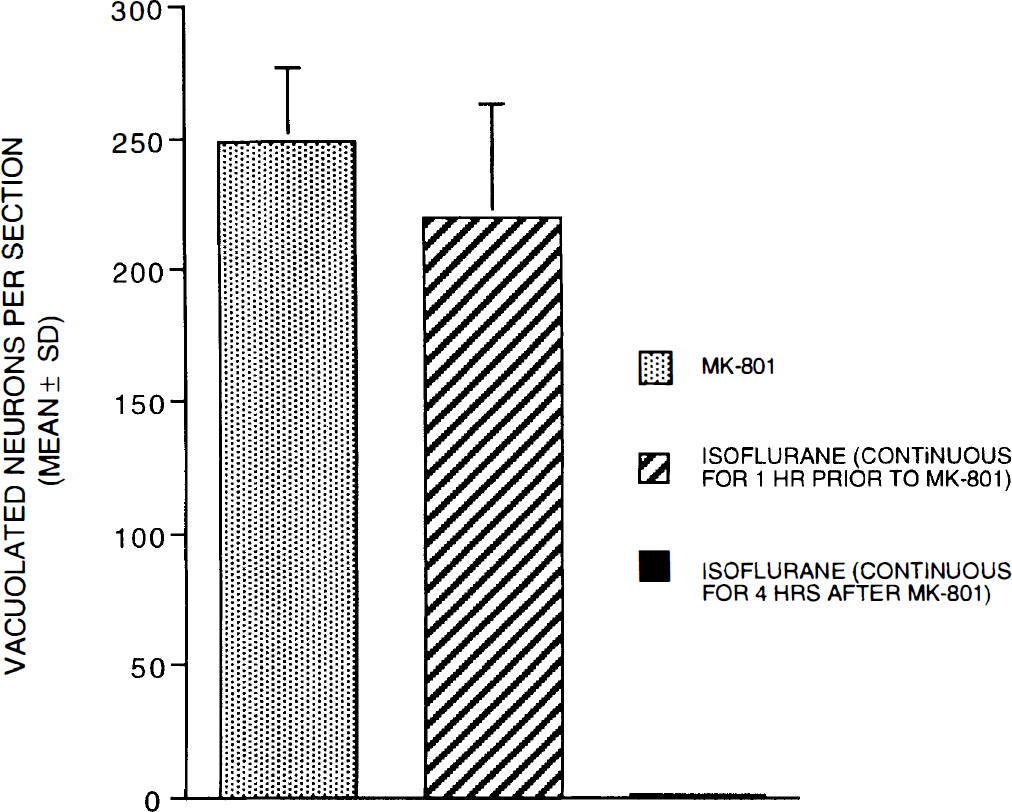
Prevention of MK-801 neurotoxicity in rat PC/RS cortex depends on the timing of isoflurane exposure. Treatment with MK-801 alone (0.5 mg/kg s.c.) caused numerous vacuolated PC/RS neurons at 4 h post-treatment. When isoflurane was administered (1.5–2.0% inhaled) for 1 h then discontinued immediately prior to MK-801, the number of vacuolated PC/RS neurons was similar to that with MK-801 alone. Co-administration of isoflurane with MK-801 during the entire 4-h period completely prevented vacuole formation (n = 8–10 rats per treatment group).
In separate experiments, isoflurane blockade of MK-801-induced vacuolization was found to be dose-dependent. When isoflurane was administered continuously from the time of MK-801 treatment to the time of death 4 h later, the minimum dose required for complete protection was 1% inhaled isoflurane (Fig. 2).
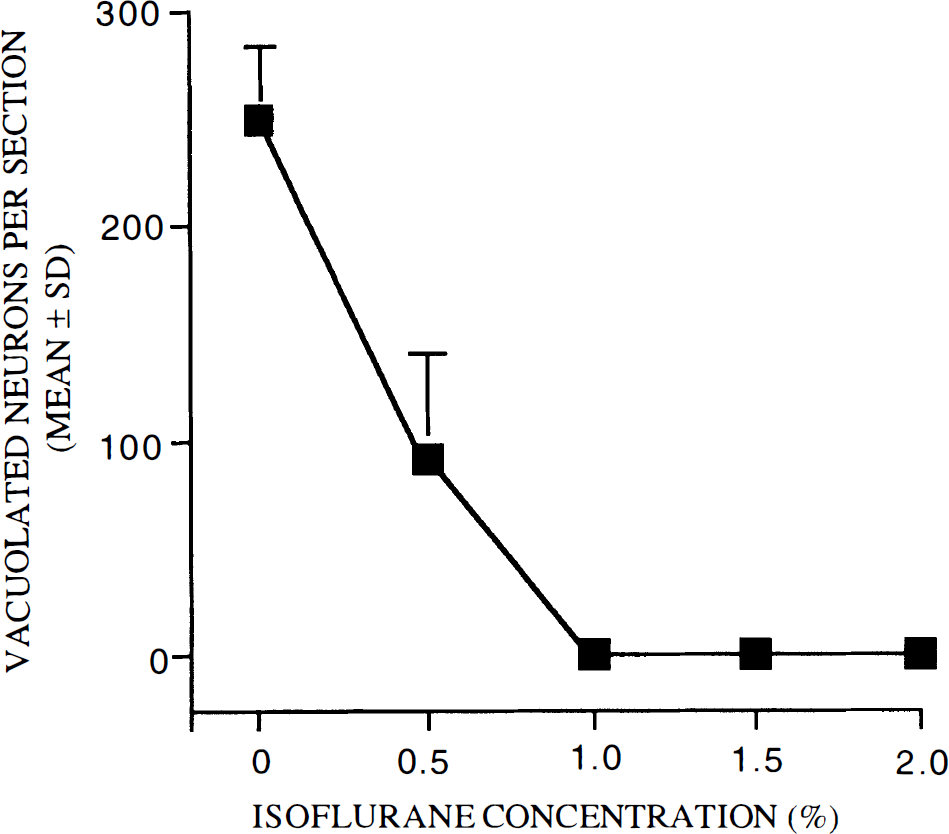
Isoflurane dose-response curve. Adult female rats were treated with a combination of MK-801 (0.5 mg/kg, s.c.) and various concentrations of inhaled isoflurane. All animals were killed 4 h later for histological evaluation of the brain. Isoflurane prevented vacuole formation in PC/RS cortex in a dose-dependent manner. The minimum dose for complete protection was 1% inhaled isoflurane (n = 8–10 rats per treatment group).
In order to assess the effects of propofol on NMDA antagonist neurotoxicity, all rats received propofol in an initial i.v. bolus dose of 20 mg/kg, then received a continuous i.v. infusion of propofol at one of several doses ranging from 5 to 20 mg/kg/h (from light sedation to general anesthesia, respectively). In these dose-response studies, all rats received MK-801 15 min after initiation of the continuous infusion of propofol and all animals were killed 4 h later for histological evaluation of the brain. At a dose of 5 mg/kg/h, the number of vacuolated PC/RS neurons was reduced to <40% that in rats treated with MK-801 alone. At doses of ≥7.5 mg/kg/h, the vacuole reaction was essentially completely prevented (Fig. 3). The histological appearance of the PC/RS cortex 4 h after MK-801 with or without propofol (20 mg/kg/h) is illustrated in Fig. 4.
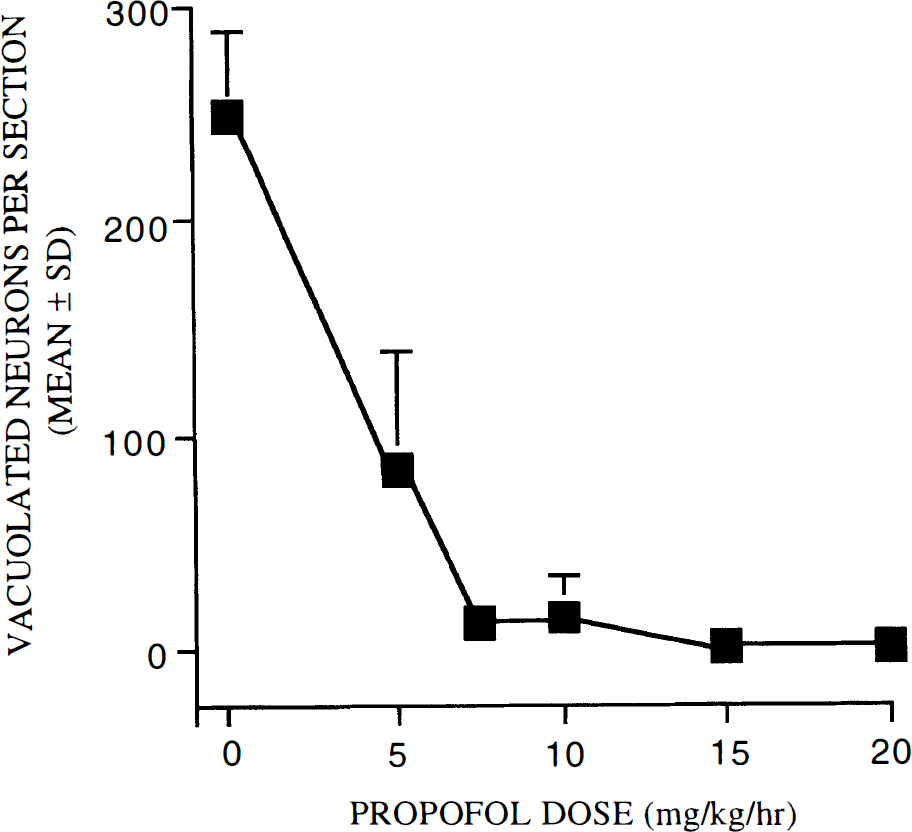
Propofol dose-response curve. A single injection of MK-801 (0.5 mg/kg s.c.) was administered together with propofol (initial bolus 20 mg/kg i.v. followed by continuous infusion at various rates from 5 to 20 mg/kg/hr). All rats were killed 4 h later for histological evaluation of the brain. Propofol was found to block vacuole formation in PC/RS cortex in a dose-dependent manner. The minimum dose for complete protection was 7.5 mg/kg/h, which is considered a low sedative dose in rats (n = 8–10 rats per treatment group).
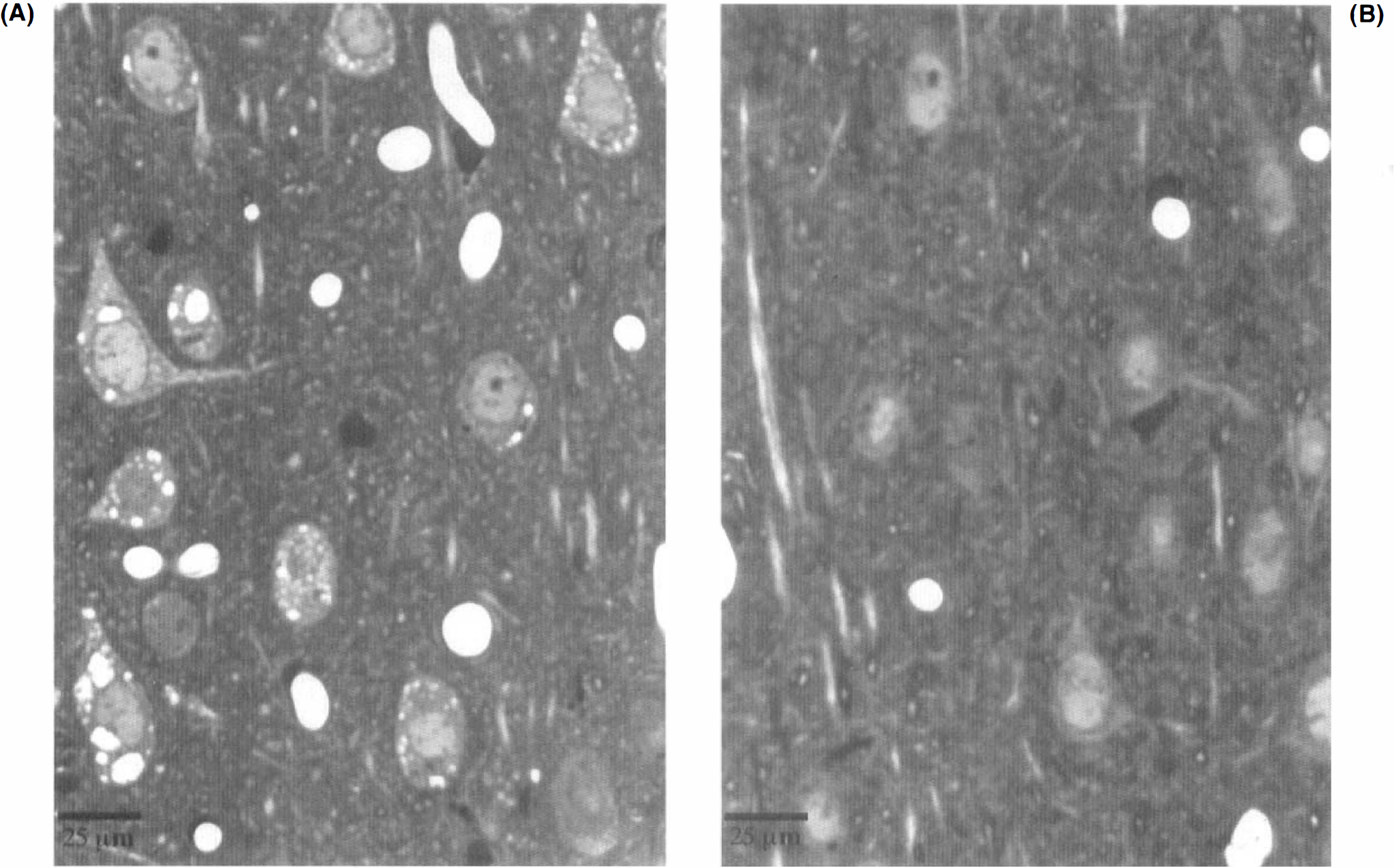
Histological appearance of the PC/RS cortex 4 h after treatment with MK-801 alone (0.5 mg/kg s.c.) (
To study the time course of propofol protection against MK-801 neurotoxicity, we first studied the time course of MK-801's neurotoxic action and found that a single dose of MK-801 (0.5 mg/kg) induces vacuolization of PC/RS neurons that is detectable in 2 h, is maximally expressed in the 4–8 h interval, and gradually declines thereafter to become essentially undetectable 18h following treatment (Fig. 5).
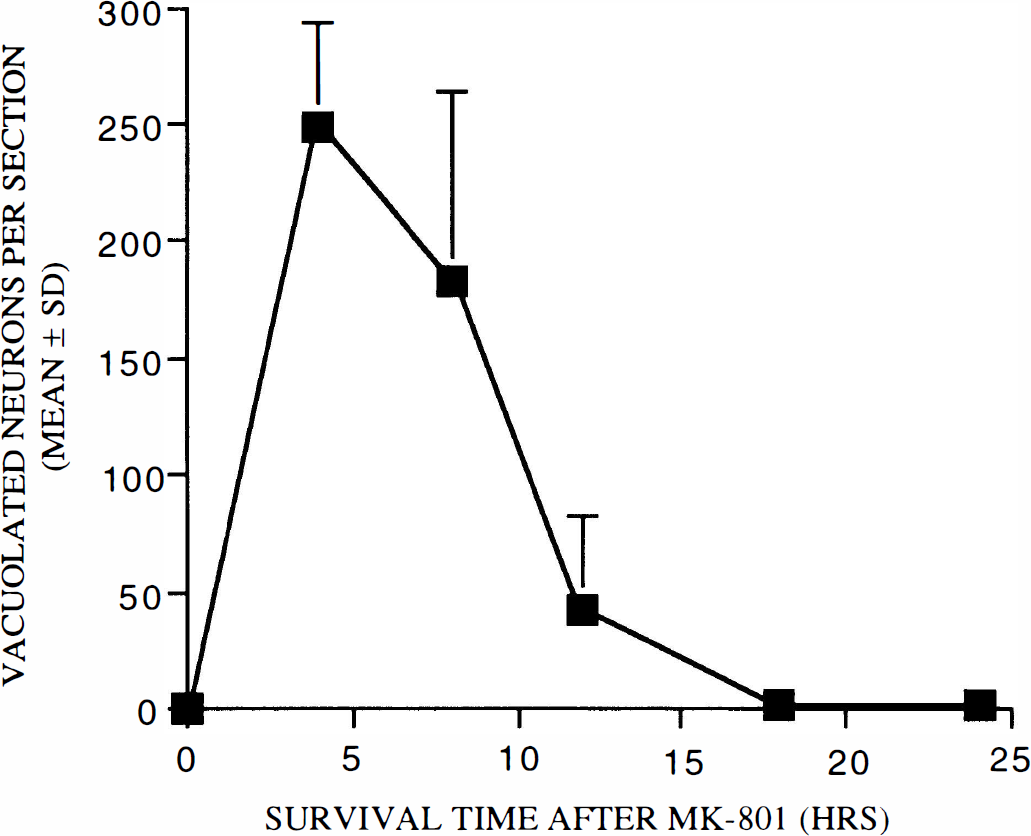
Time course of MK-801 neurotoxicity. A single dose of MK-801 (0.5 mg/kg s.c.) induces vacuolization of PC/RS neurons that is maximally expressed after 4 h and gradually declines over the following 8 h to become undetectable at 18 h (n = 8–10 rats per treatment group).
This time course profile for MK-801 neurotoxicity suggested the possibility that blockade of the neurotoxic process for the first 4 h after MK-801 treatment might not be adequate to completely prevent the neurotoxic reaction. Additional experiments were undertaken to address this issue. The standard s.c. dose of MK-801 (0.5 mg/kg) was co-administered with an i.v. bolus (20 mg/kg) of propofol followed by continuous propofol infusion of 20 mg/kg/h for 4 h. Animals were then killed at 4, 8, and 12 h from the time of MK-801 treatment. Brains examined at 4 h (immediately after stopping the propofol infusion) had no PC/RS vacuolated neurons (Fig. 6). However, brains examined at 8 h (4 h after stopping the propofol infusion) showed only a partial protective effect (59% as many vacuolated neurons as MK-801 controls). Animals that lived 8 h after stopping the propofol infusion had the same number of vacuolated neurons as did MK-801 controls, but this merely reflected the fact that the reaction in both experimental and control animals had largely subsided by this time.
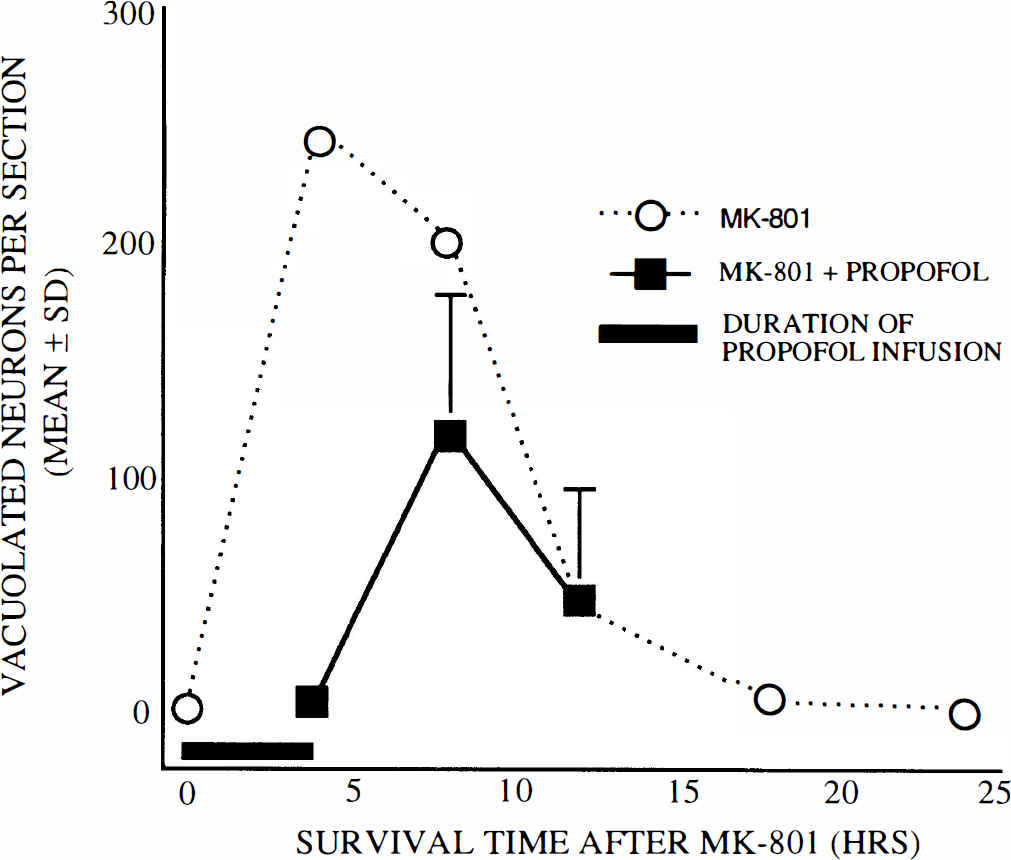
Effects of propofol (20 mg/kg/h) on MK-801 neurotoxicity. Rats were treated with MK-801 alone (dotted lines) or with MK-801 plus an i.v. propofol infusion (20 mg/kg bolus followed by 20 mg/kg/h) for 4 h. Animals killed immediately after the propofol infusion was stopped had no PC/RS vacuolated neurons. If the animal was killed 4 h after the infusion was stopped, the vacuolated neuron count was 59% of that for MK-801 controls; if the animal was killed 8 h after the infusion was stopped, the vacuolated neuron count was the same as for MK-801 controls (n = 8–10 rats per treatment group).
In an additional experiment, we attempted to determine how much protection could be achieved by increasing the duration of propofol infusion to 8 h with the same dose of propofol (20 mg/kg/h) utilized in the previous experiment. However, this dose was lethal to a high percent of animals in the 5–7 h-infusion interval. Therefore, we conducted these experiments with lower doses of propofol (7.5 and 10 mg/kg/h). When propofol was infused for 8 h at 7.5 mg/kg/h and the brains examined immediately, there were 17% as many vacuolated neurons as in controls (Fig. 7). If the animals were killed 4 h later (12 h after MK-801), there were ∼60% as many vacuolated neurons as in corresponding control brains. When propofol was infused for 8 h at 10 mg/kg/h and the brains examined immediately, there were no vaculated neurons; if animals were killed 4 h later (12 h after MK-801), there were ∼60% as many vacuolated neurons as in corresponding controls (Fig. 8). In an additional experiment, propofol was infused for 12 h at 10 mg/kg/h and the animals killed immediately or 6 h later; no vacuolated neurons were found at either time (Fig. 9).
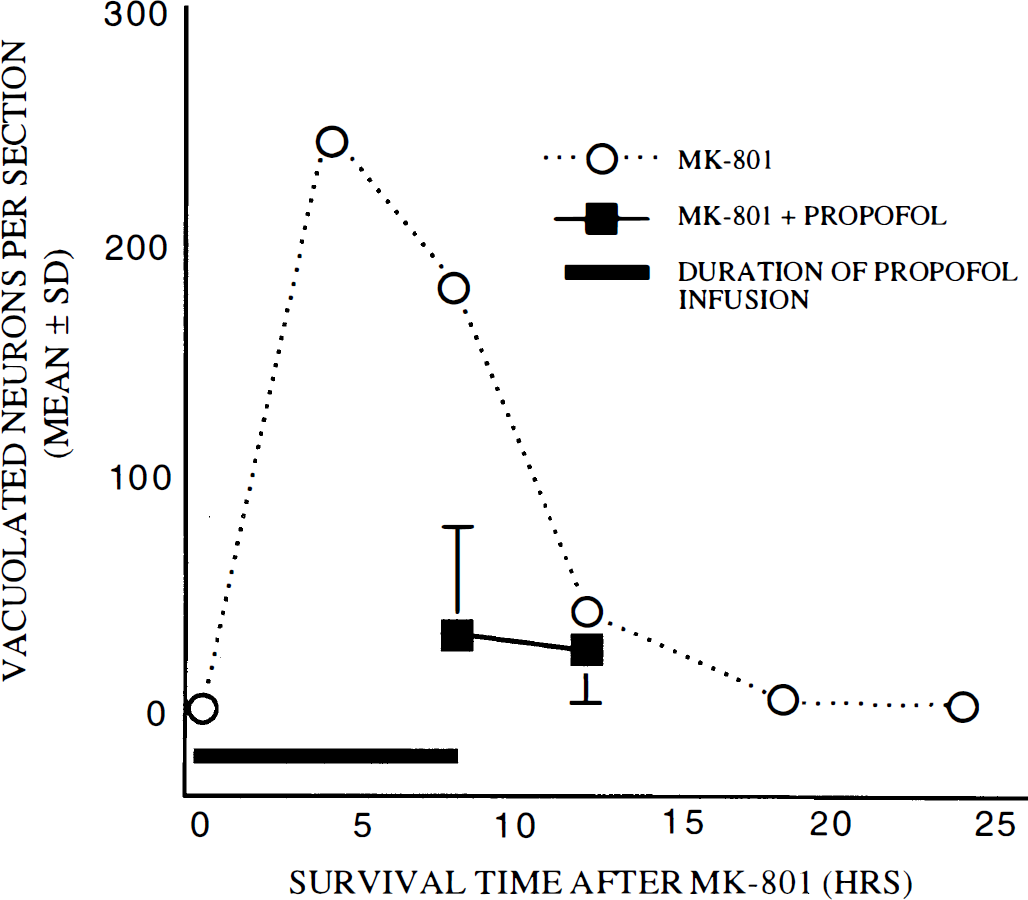
Propofol infusion (7.5 mg/kg/h i.v.) for 8 h. Rats were treated with MK-801 alone (dotted lines) or with MK-801 plus an i.v. propofol infusion (20 mg/kg bolus followed by 7.5 mg/kg/h i.v.) for 8 h. Animals killed immediately after the propofol infusion was stopped had ∼17% as many vacuolated neurons as did MK-801 controls. If the animal was killed 4 h after the infusion was stopped (12 h after MK-801 treatment), the vacuolated neuron count was 60% of that for corresponding MK-801 controls (n = 8–10 rats per treatment group).
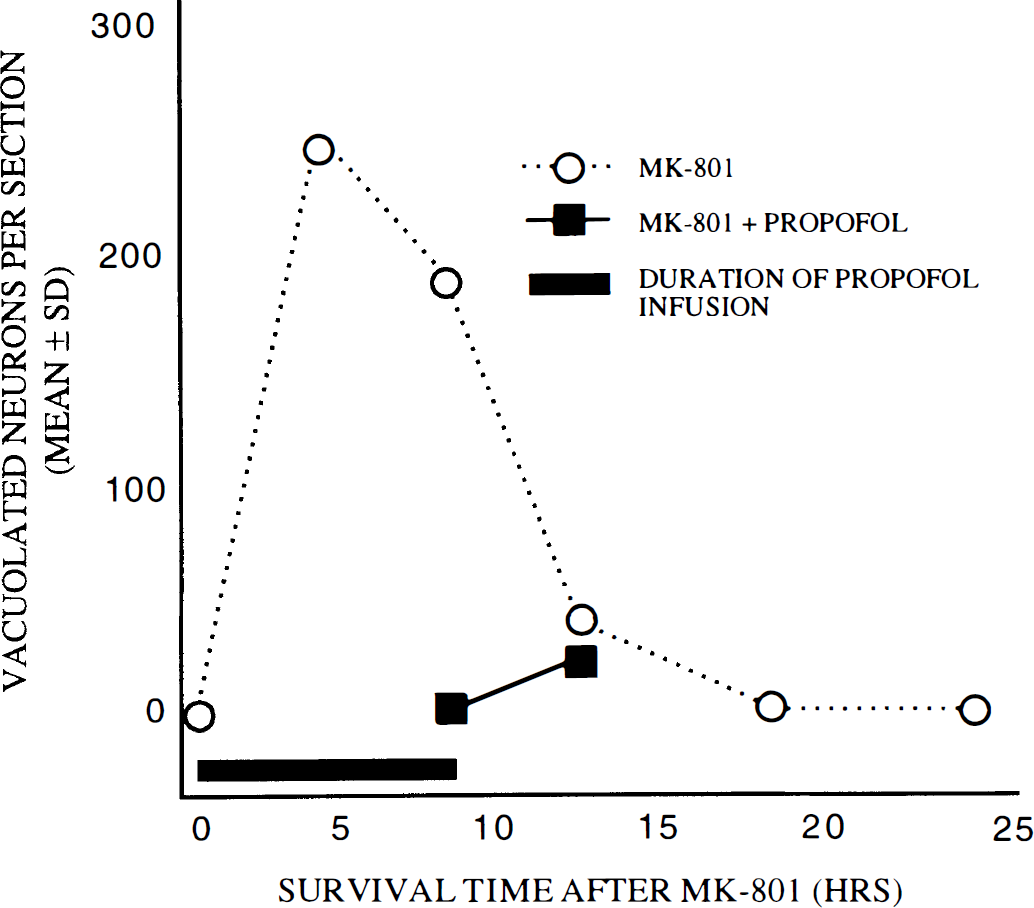
Propofol infusion (10 mg/kg/h i.v.) for 8 h. Rats were treated with MK-801 alone (dotted lines) or with MK-801 plus an i.v. propofol infusion (20 mg/kg bolus followed by 10 mg/kg/h i.v.) for 8 h. Animals killed immediately after the propofol infusion was stopped had no vacuolated neurons. In animals killed 4 h after the infusion was stopped (12 h after MK-801 treatment), the vacuolated neuron count was 60% that for the corresponding MK-801 controls (n = 8–10 rats per treatment group).
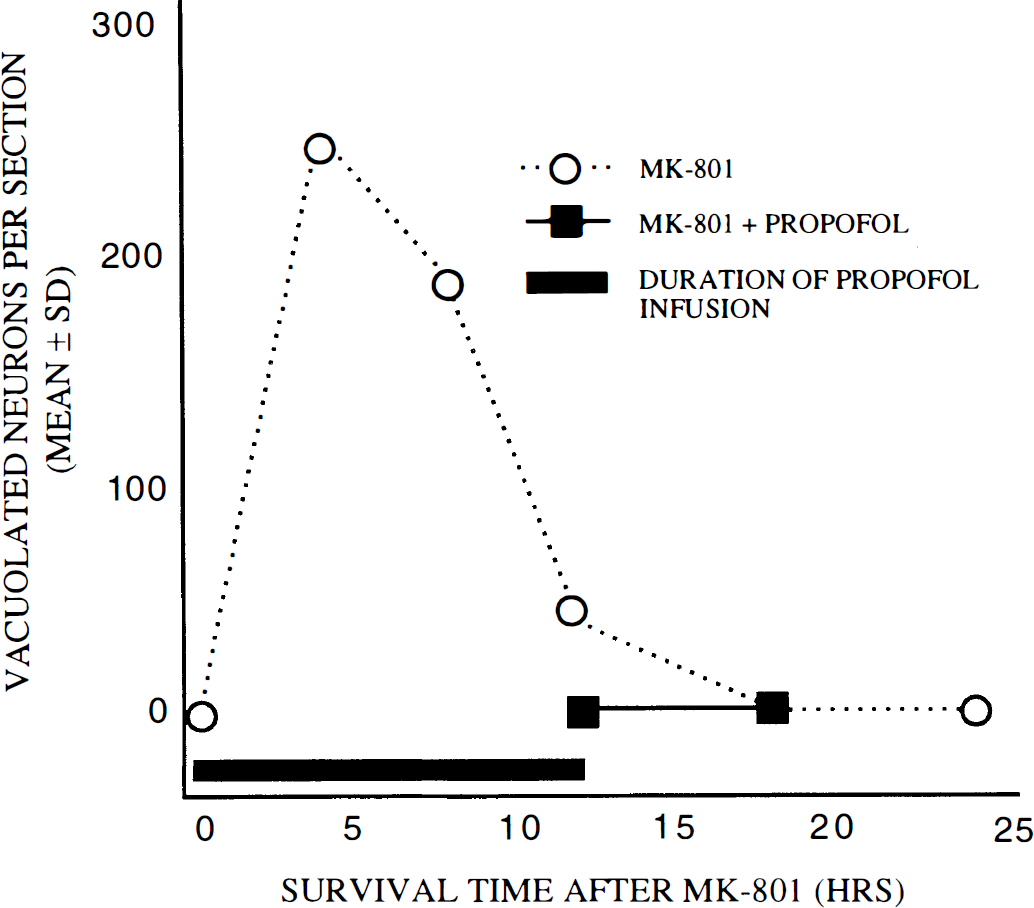
Propofol infusion (10 mg/kg/h i.v.) for 12 h. Rats were treated with MK-801 alone (dotted lines) or with MK-801 plus an i.v. propofol infusion (20 mg/kg bolus followed by 10 mg/kg/h i.v.) for 12 hrs. Animals killed either immediately after the propofol infusion was stopped or 6 h later had no vacuolated neurons (n = 8–10 rats per treatment group).
When 7.5 or 10 mg/kg/h infusion rates were used, animals appeared sedated but had positive corneal and eye lid reflexes, and readily responded to paw pinch. After discontinuation of the infusion, animals were fully awake within 20 min and had a positive righting reflex.
DISCUSSION
In these experiments, we demonstrated that two widely used general anesthetics, isoflurane and propofol, can prevent the powerful NMDA antagonist, MK-801, from injuring cerebrocortical neurons. In recent years, we have been intensively studying the mechanism underlying MK-801 neurotoxicity. There is extensive evidence (reviewed in Olney and Farber, 1995) that this type of neurotoxicity is mediated by a disinhibition mechanism in which GABAergic neurons are inactivated by blockade of the NMDA receptors that are present on these neurons. An injurious excitotoxic process is thereby unleashed upon cerebrocortical neurons. Drugs that act at GABAA receptors to restore inhibitory tone (e.g., benzodiazepines, barbiturates, and halothane) prevent unleashing of excitotoxic activity and thereby protect cerebrocortical neurons from inury (Olney et al., 1991; Ishimaru et al., 1995). We propose that this is the mechanism that mediates the neuroprotective actions of isoflurane and propofol since both agents are known to act at GABAA receptors to augment GABAergic neurotransmission (Nakahiro et al., 1989; Hara et al., 1993; Franks and Lieb, 1994).
Our data indicate that in order to completely prevent expression of MK-801 neurotoxicity, isoflurane or propofol must be administered continuously for 8–12 h after MK-801 administration, i.e., for the entire period of time that MK-801 is active in the brain. This is because the duration of action of these anesthetic agents in the brain is very brief compared to the very long duration of action of MK-801 (Adams et al., 1980; Hucker et al., 1982; Stevens and Kingston, 1992).
Although MK-801 is the agent used in most animals experiments pertaining to NMDA antagonist neurotoxicity, it is known that most, if not all, NMDA antagonists that effectively block passage of ion currents through NMDA receptors mimic the neurotoxic action of MK-801. For example, all agents that act at the phencyclidine (PCP) recognition site within the NMDA receptor ion channel (PCP, MK-801, ketamine, and tiletamine) and those that act at the glutamate recognition site outside the NMDA ion channel (CPP, CPP-ene, and CGS 19755) induce this neurotoxic reaction (Olney et al., 1989; Olney et al., 1991; Hargreaves et al., 1993). In addition, although PCP and ketamine are the NMDA antagonists most extensively studied for their psychotomimetic actions (Domino and Luby, 1962; Reich and Silvay, 1989), recent human clinical trials with CPP, CPP-ene, and CGS 19755 have revealed that all three of these agents trigger the same type of psychotic reactions as do PCP and ketamine (Kristensen et al., 1992; Herrling, 1994; Grotta, 1994; Muir and Lees, 1995). In view of evidence from human studies that GABAergic agents (benzodiazepines and barbiturates) suppress the psychotomimetic actions of NMDA antagonists (Magbagbeola and Thomas, 1974; Reich and Silvay, 1989), it seems likely that isoflurane and propofol may also have this beneficial property. Thus, if stroke or brain trauma patients were anesthetized with these agents while they are being treated with an NMDA antagonist, they would not only be protected against the cerebrocortical injurious effects of the NMDA antagonist but also psychotic reactions. The patient would sleep comfortably while under NMDA antagonist treatment and then would awaken without experiencing an “emergence” psychosis.
Although isoflurane is very effective in controlling MK-801 neurotoxicity and could easily be utilized in the operating room, the complicated setup required for isoflurane administration may preclude its use in an intensive care setting (e.g., for management of stroke or brain trauma); in these situations, propofol might be ideal since it has been widely used for long-term sedation in critically ill patients and offers quick onset of action and prompt recovery after continuous infusion is discontinued (Beller et al., 1988; Bailie et al., 1992). The level of anesthesia could be controlled from very light sedation to deep sleep. Our study indicates that sedative doses of propofol are as effective as high (general anesthetic) doses in blocking neurotoxicity. The doses used in the present experiments are substantially higher than would be used in a human clinical setting due to the interspecies differences in the pharmacokinetics of propofol. For example, an anesthetic dose of propofol is 3–6 mg/kg/h in humans (Fragen and Avram, 1992) whereas 60 mg/kg/h is required to achieve a comparable level of anesthesia in the rat (Shyr et al., 1995; Yang et al., 1995).
Unlike halothane, which is contraindicated for acute brain injury patients because it increases intracranial pressure, isoflurane and propofol are considered favorable for such patients (Madsen et al., 1987; Pinaud et al., 1988). Both agents decrease the cerebral metabolic rate and cerebral metabolic requirements for oxygen. In addition, propofol causes cerebral vasoconstriction and thereby decreases intracranial pressure (Herregods et al., 1989; Plainer et al., 1989; Weinstabl et al., 1990). If given in high doses, propofol induces an isoelectric state in the brain (so-called burst suppression) (Veselis et al., 1991) and is commonly used in neurosurgery for brain protection. Unlike most GABAergic agents (Alquander, 1978), propofol has a very short duration of action that allows rapid recovery from anesthesia and early warning of mental status deterioration (Cockshott, 1985); in addition, propofol has been administered by continuous i.v. infusion to humans for up to several weeks without inducing tolerance, physical dependence, or withdrawal reactions (Borgeat et al., 1994).
Recent studies (Yamakura et al., 1995) suggest that propofol, in addition to its action at GABAA receptors, may exert a weak blocking action at NMDA receptor channels as expressed in Xenopus oocytes. Thus, if used together with another more potent NMDA antagonist in the treatment of stroke or brain trauma, propofol may enhance the degree of NMDA receptor blockade, thereby maximizing neuroprotection while preventing both the neurotoxic and psychotomimetic side effects that would otherwise accompany such blockade.
In conclusion, isoflurane and propofol may be suitable agents for use with an NMDA antagonist to enable the NMDA antagonist to be used more safely in the therapeutic management of acute brain injury syndromes.
Footnotes
Acknowledgment:
This research was supported in part by a National Institute of Mental Health Research Scientist Award MH 33894 (J.W.O.) and National Institutes of Health grants EY 08089, DA 05072, and AG 11355. The authors would like to thank Dr. David Wozniak for valuable discussions, and Joann Labruyere, Tony Nardy, and Sarah K. Eye for technical help.