
Abstract
Several lines of inquiry have indicated that glycine plays an important role in both glutamatergic neurotransmission and pathophysiology of cerebral ischemia. However, subacute outcome trials demonstrating the efficacy of glycine antagonists as neuroprotectants have not been performed with rigorous control of brain temperature. In this study, we investigated the effect of N-methyl-D-aspartate (NMDA) receptor glycine recognition site antagonism in a temperature-controlled rodent model of transient focal ischemia. Male Wistar rats underwent 75 min of intraluminal middle cerebral artery occlusion (MCAO). During MCAO and the first 24 h of reperfusion, rats (n = 10) were administered either vehicle or the glycine antagonist 5-nitro-6,7-dichloro-2,3-quinoxalinedione (ACEA 1021) i.v. as a bolus infusion of 5 mg/kg followed by 3.5 mg/kg/h (Low-Dose) or 10 mg/kg followed by 7 mg/kg/h (High-Dose) for 24 h. Cortical temperature was controlled at 38.0 ± 0.1°C during MCAO and the first 6 h of reperfusion. A 7-day recovery interval was allowed. Mean total infarct volume was reduced by ∼ 40% in both high- and low-dose groups (p < 0.01). The preponderance of infarct reduction occurred in the cortex (p < 0.01). Neurologic function correlated with the size of cerebral infarct (p = 0.001). Neurologic grade was similarly improved by treatment with either dose (p = 0.01). These results demonstrate that neuroprotection achieved by antagonism of the glycine recognition site persists when brain temperature is controlled, indicating a potent mechanism of action other than attenuating a hyperthermic response to ischemia.
A theoretical pharmacologic approach to amelioration of ischemic brain damage is antagonism of the glycine recognition site of the N-methyl-
Recently, several compounds have been synthesized that are potent and selective antagonists of the strychnine-insensitive glycine receptor. In laboratory models of either ischemia or head trauma, a reduction in injury has been observed following use of these drugs (Gill et al., 1995; Newell et al., 1995; Takano et al., 1996; Tsuchida and Bullock, 1995; Warner et al., 1995b). Our previous work with one such compound, 5-nitro-6,7-dichloro-2,3-quinoxalinedione (ACEA 1021) (Woodward et al., 1995), examined infarct volume in awake rats undergoing intraluminal middle cerebral artery occlusion (MCAO) (Warner et al., 1995b). Although marked protection was observed, brain temperature was not controlled. Rectal temperature was monitored and a clear dose-dependent effect of the drug in inhibiting a hyperthermic response to ischemia was observed.
Numerous investigations have shown that the extent of ischemic brain damage is influenced by brain temperature during the ischemic interval (Busto et al., 1989; Chen et al., 1993; Colbourne and Corbett, 1995; Ridenour et al., 1992). In the intraluminal MCAO model, it has been shown that ischemia induces a transient episode of hyperthermia (Kuluz et al., 1993; Zhao et al., 1994). Hyperthermic temperature increases of as little as 1.2°C have been shown to nearly double the size of the resultant infarct (Warner et al., 1993). Therefore, the possibility is real that the previously reported protective effects of glycine recognition site antagonism represent only temperature-reducing effects of this class of compounds. Accordingly, a study was designed to examine this issue.
METHODS AND MATERIALS
This study was approved by the Duke University Animal Care and Use Committee. Male Wistar rats (aged 8–10 weeks) were anesthetized with 40 mg/kg i.p. sodium pentobarbital (Nembutal, 50 mg/ml, Abbott Laboratories, North Chicago, IL, U.S.A.). Each animal was positioned in a stereotactic head frame. A midline scalp incision was made. One mm lateral to midline, a burr hole was drilled over the left hemisphere at bregma = +2 mm. A radiotelemetry thermistor (Brain Probe, model XM-FH, Mini Mitter, Inc. Sunriver, OR, U.S.A.) was advanced into the cerebral cortex to a depth of ∼2 mm. The probe was fixed in place with two cranial screws and orthodontic resin. The wound was closed with suture and animals were returned to their cages after recovery of the righting reflex.
Prior to this procedure, the temperature probe was calibrated in a circulating water bath against a mercury thermometer within the range of 35.0–40.0°C. This allowed extrapolation of temperatures from calibration points in accordance with the radiofrequency emitted by the thermistor. Radiofrequency signals from the radiothermistor were received (Telemetry Receiver Model RA1010, Data Science, St. Paul, MN, U.S.A.), converted to digital signals, and processed through a computer (4DX-33V, Gateway 2000, North Sioux City, SD, U.S.A.) with software written to allow monitoring and automated control of cortical temperature.
At 2–4 days following the above procedure, rats were fasted from food for 12–14 h but allowed free access to water. They were then anesthetized with 1–2% halothane in 50% O2/balance N2, (vol/vol) delivered via snout mask, and allowed to breathe spontaneously. Via incision, a catheter was placed into the tail artery to monitor MABP and to sample blood. Via a lateral cervical skin incision, a catheter was placed into the left external jugular vein. This catheter was filled with saline, tunneled subcutaneously, and exteriorized on the nape of the neck.
Animals were then prepared for middle cerebral artery occlusion (MCAO) using modifications of techniques described by others (Memezawa et al., 1992; Zea Longa, et al., 1989). Via a right lateral cervical skin incision, the right common carotid artery (CCA) was identified. The external carotid artery (ECA) was isolated, and the occipital, superior thyroid, and external maxillary arteries ligated and divided. The internal carotid artery (ICA) was then dissected distally until the origin of the pterygopalatine artery was visualized. Following surgical preparation, a 20-min interval was allowed for physiological stabilization. During this interval, arterial blood was sampled to measure pH, PAO2, PaCO2, plasma glucose, and hematocrit.
Rats then underwent 75 min of MCAO. A 0.25 mm diameter nylon filament was inserted into the ECA stump and advanced ∼23 mm into the ICA to occlude the middle carotid artery (MCA). Immediately after filament insertion, the neck wound was closed with suture and halothane was immediately discontinued allowing the animal to promptly awaken. Animals were then placed in a 10-L Plexiglas box containing 50% O2/50% N2 (vol/vol).
Animals were randomly assigned to one of three groups: (a) Vehicle (n = 10). Bolus i.v. infusion (4.2 ml/kg over 1 min) of ACEA 1021 vehicle (0.05 M Tris in H2O) followed by a continuous 24-h i.v. infusion of vehicle at a rate of 2.9 ml/kg/h; (b) Low-Dose (n = 10). Bolus i.v. infusion (5.0 mg/kg over 1 min) of ACEA 1021 (CoCensys, Inc., Irvine, CA, U.S.A.) (2.4 mg/ml) followed by a continuous 24 hr i.v. infusion of ACEA 1021 at a rate of 3.5 mg/kg/h; and (c) High-Dose (n = 10). Bolus i.v. infusion (10.0 mg/kg over 1 min) of ACEA 1021 followed by a continuous 24-h i.v. infusion of ACEA 1021 at a rate of 7.0 mg/kg/h.
Onset of bolus and continuous infusion commenced 10 min after onset of ischemia. ACEA 1021 was dissolved in 0.05 M Tris in H2O at a concentration of 2.4 mg/ml. Volumes of vehicle administered to the Vehicle group were intended to equal drug volume administered to the High-Dose group.
MABP was measured continuously throughout the ischemic interval. Arterial blood gases and pH were determined 45 min after onset of ischemia. At termination of ischemia, halothane was introduced into the respiratory gas mixture. Rats were then allowed to breath spontaneously with ∼1% halothane administered in 50% O2/50% N2 (vol/vol) via snout mask to facilitate removal of the filament. The filament was removed and the cervical wound closed with sutures. At termination of ischemia, arterial blood was collected for later measurement of ACEA 1021 plasma concentration. The arterial catheter was then removed.
At 10 min after filament removal, halothane was discontinued and the rats were allowed to awaken. Rats were returned to the Plexiglas box for an additional 6 h of temperature control. Brain temperature was controlled as follows. During MCAO and the first 6 h of reperfusion, cortical temperature was regulated at a target temperature of 38.0°C. If the cortical temperature was <38°C, a heat lamp was automatically turned on. If cortical temperature was > 38.0°C, chilled room air was blown over the surface of the animal by automated switching of an air-line solenoid valve.
At 24 h after filament removal, the respective infusion regimens were discontinued. Rats were administered ∼1% halothane in 50% O2/50% N2 (vol/vol) via snout mask. The jugular venous catheter was surgically removed. After recovery, animals were returned to their cages for the remainder of the 7-day postischemic recovery interval.
All animals were evaluated neurologically 7 days after reperfusion. Each rat was assigned a score of 0–3, where 0 = no observable deficit, 1 = forelimb flexion, 2 = decreased resistance to lateral push without circling, and 3 = same behavior with circling (Bederson et al., 1986). Neurological examination was performed by one observer blinded to group assignment.
Following neurological evaluation, animals were weighed, anesthetized with halothane, and killed by decapitation. The brain was removed and frozen at −20°C in 2-methylbutane. Using a cryotome, quadruplicate 20 μm thick coronal sections were taken at 660 μm intervals over the rostral–caudal extent of the infarct. Sections were dried and stained with hematoxylin and eosin.
Infarct volume was measured by digitally sampling stained sections with a Sony CCD Model XC-77 video camera (Sony, Japan) controlled by an image analyzer (M2 Turnkey System, Imaging Research, Inc., St. Catharines, Ontario, Canada). The image of each section was stored as a 512 × 512 matrix of 14 μm2 pixel units. The digitized image was then displayed on a video screen. With the observer blinded to experimental condition, infarct borders in both cortex and subcortex were individually outlined (corpus callosum excluded) using an operator-controlled cursor. The area (mm2) of infarct was determined automatically by counting pixels contained within the outlined regions of interest (ROIs). Infarct volumes (mm3) were computed as running sums of infarct area multiplied by the known interval (i.e., 660 μm) between sections over the extent of the infarct, expressed as an orthogonal projection.
Plasma samples drawn for ACEA 1021 analysis were vortex-mixed with an equal volume of 20% (vol/vol) trichloroacetic acid (Fisher Scientific, Fairlawn, NJ, U.S.A.) to precipitate plasma proteins. The mixture was centrifuged for 5 min at 12,000 g. The supernatant was filtered through a 0.45-mm nylon Acrodisc HPLC certified filter (Gelman, MI, U.S.A.). A 10-μl aliquot sample was injected onto a Vydac RPC C18 HPLC column and eluted with a mobile system consisting of 0.1% trifluoroacetic acid (TFA) and a 20–100% gradient of acetonitrile containing 0.1% TFA. Absorbence was monitored with an ultraviolet (UV) detector at 217 nm. The HPLC was a Beckman System Gold (Fullerton, CA, U.S.A.). Prior to analysis of samples, a standard curve was generated that spanned the range of expected concentrations of ACEA 1021.
Physiologic values were compared qualitatively between groups to preserve statistical power for analysis of major dependent variables. Infarct volumes were compared between groups by one-way analysis of variance (ANOVA). Infarct volumes were correlated with neurologic grades by the Spearman rank correlation coefficient while neurologic grades were compared between groups by the Kruskal– Wallis H statistic. Parametric values are expressed as mean ± SD.
RESULTS
Physiologic values for the three experimental groups are given in Table 1. No substantive inter-group differences were noted for MABP, arterial blood gases/pH, plasma glucose, hematocrit, or brain temperature. Preischemic hypercapnia reflects spontaneous ventilation allowed to occur during halothane anesthesia. This resolved with discontinuation of the anesthetic. Animals had fully recovered to a state of normocapnia and mild hyperoxemia as documented by blood gas analysis 45 min after onset of ischemia. Neither dose of ACEA 1021 appeared to alter ventilation. Computer-assisted servocontrol of temperature during ischemia and the first 6 h after reperfusion was successful in maintaining strict regulation of cortical temperature within ±0.1°C of the set-point (38.0°C). ACEA 1021 plasma concentrations measured immediately prior to filament removal were 28 ± 7 μg/ml (Low-Dose group) and 56 ± 4 μg/ml (High-Dose group). No ACEA 1021 was detected in plasma from vehicle-treated animals.
Physiologic values for the experimental groups
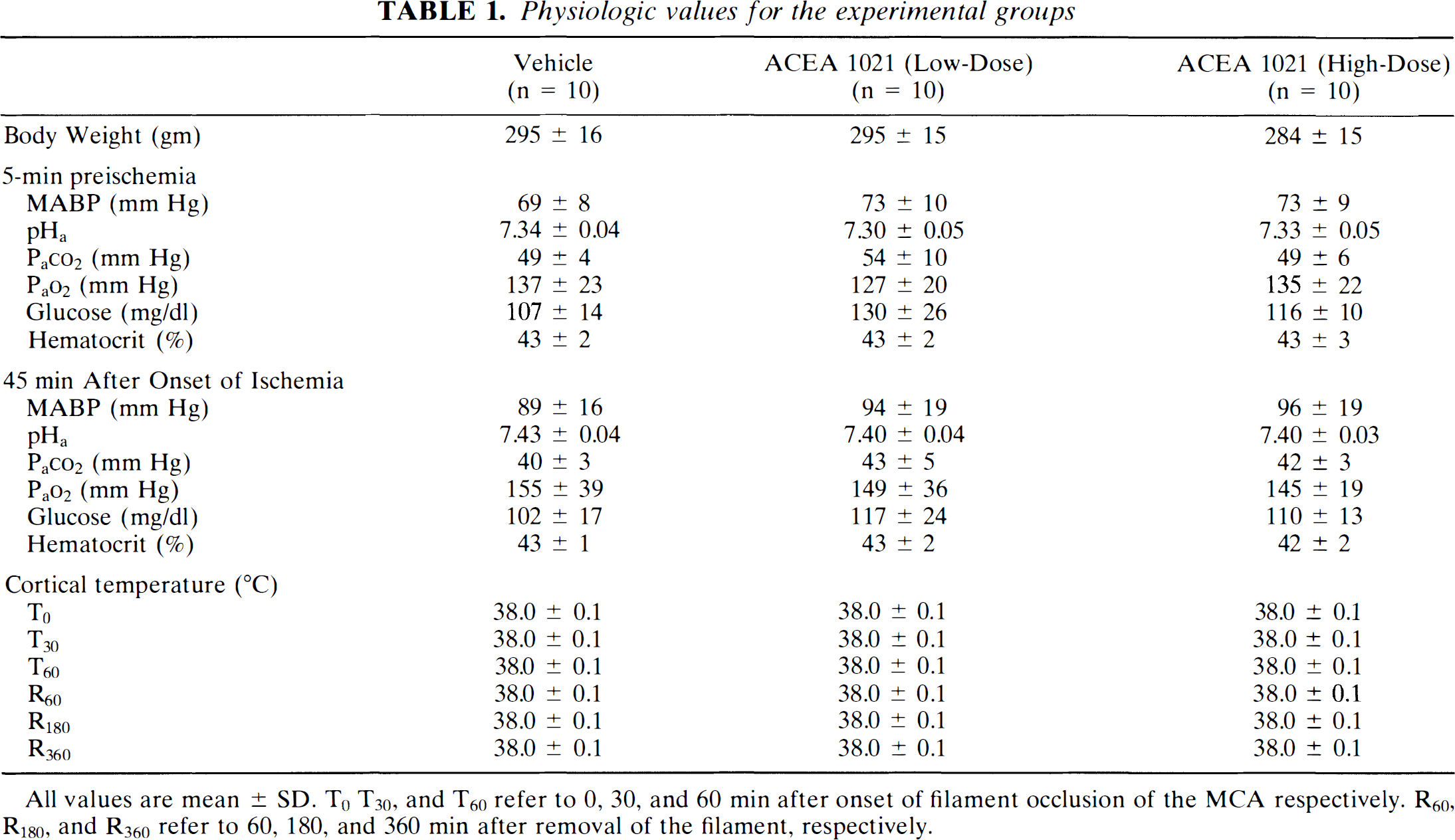
All values are mean ± SD. T0 T30, and T60 refer to 0, 30, and 60 min after onset of filament occlusion of the MCA respectively. R60, R180, and R360 refer to 60, 180, and 360 min after removal of the filament, respectively.
Glycine recognition site antagonism appeared to have a sedative effect, particularly when combined with halothane. Vehicle-treated animals typically regained a standing posture within 15 ± 7 after discontinuation of halothane at onset of ischemia. In contrast, for the Low- and High-dose groups, this occurred within a 34 ± 12 and 45 ± 9 min, respectively. A similar pattern was observed after recovery from halothane anesthesia at the termination of ischemia. However, after effects of halothane had dissipated, all rats remained ambulatory and readily responsive to auditory and visual stimuli throughout the postischemic infusion period.
Total cerebral infarct volumes are presented in Fig. 1. A main effect for each treatment group was observed (p = 0.001). Both ACEA 1021 treatment regimens produced a similar but significant (p < 0.01) reduction in total infarct volume within the range of 40–45% versus vehicle. Cortical infarct volume was significantly reduced by both doses of drug (Vehicle = 76 ± 16 mm3; Low-Dose = 30 ± 37 mm3; High-Dose = 38 ± 33 mm3, p < 0.01). Although a trend was present (Vehicle = 76 ± 18 mm3; Low-Dose = 57 ± 30 mm3; High-Dose = 53 ± 19 m3), ACEA 1021 failed to significantly reduce subcortical infarct volume (p = 0.07).
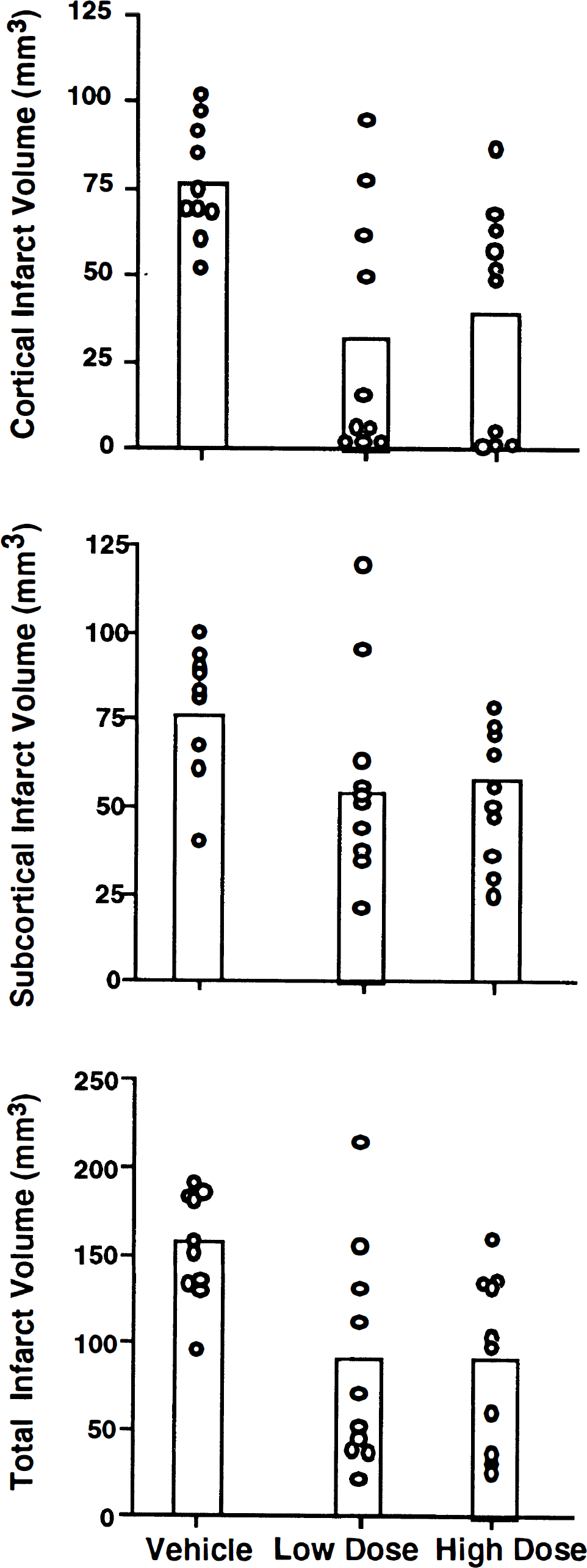
Cortical, subcortical, and total cerebral infarct volumes for individual rats are depicted. Boxes indicate mean values for each group. Low- and High-Dose groups designate, respectively, rats that received the glycine recognition site antagonist ACEA 1021 i.v. either as a 5 mg/kg bolus followed by a 24-h infusion at 3.5 mg/kg/h or a 10 mg/kg bolus followed by a 24-h infusion at 7 mg/kg/h. Significant reductions in cortical and total infarct volume were observed in the Low- and High-Dose groups (p < 0.01) relative to the Vehicle group.
There was no evidence of grand mal type seizure activity in any animal. A few rats were noted to have hemifacial spasm ipsilateral to the lesion during the 6-h recovery interval. This was not associated with treatment group. There were no stroke related deaths. Neurologic function correlated with the size of cerebral infarct (p = 0.001), although circling was not uniformly present in animals with larger infarcts. Neurologic grade was improved by treatment with either dose of the glycine recognition site antagonist (p = 0.01) (Fig. 2).
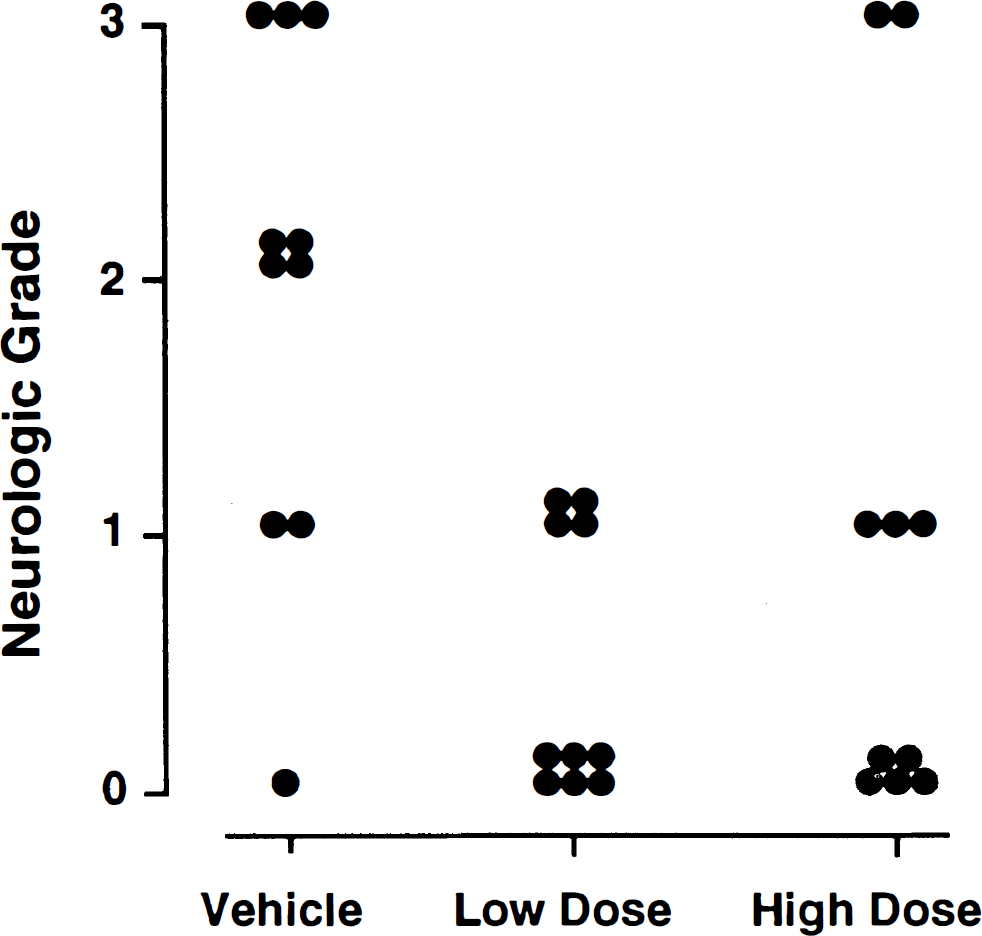
At 7 days after 75 min of intraluminal occlusion of the MCA, rats were examined for absence (0) or presence (3) of severe hemiparesis. Each point depicts values for a single rat. Low- and High-Dose groups designate, respectively, rats that received ACEA 1021 i.v. either as a 5 mg/kg bolus followed by a 24-h infusion at 3.5 mg/kg/h or a 10 mg/kg bolus followed by a 24-h infusion at 7 mg/kg/h. Antagonism of the NMDA receptor glycine recognition site resulted in significant improvement in neurologic grade (p = 0.01) at either dose when compared to vehicle.
DISCUSSION
Results of this experiment indicate that NMDA receptor glycine recognition site antagonism effectively attenuates injury resulting from a temporary focal cerebral ischemic insult. This occurred independently of effects on hemodynamics, ventilation, plasma glucose concentration, or thermoregulation. Neuroprotection persisted through at least 7 days postinsult. Prior work with a temperature-unregulated filament occlusion model showed ∼75% reduction of mean cortical infarct volume by ACEA 1021 (Warner et al., 1995b). In the present study, a ∼60% reduction in mean cortical infarct size was observed, indicating the robust nature of glycine recognition site antagonism as a strategy for neuroprotection even within a temperature-regulated model.
Antagonists of the glycine recognition site have received increasing attention as potential pharmacologic approaches to treatment of ischemic/traumatic brain injury for several reasons. To date, adverse psychotomimetic effects of these compounds have not been observed, a potential distinct advantage over both competitive and noncompetitive NMDA receptor antagonists (Balster et al., 1995; Grotta et al., 1995). As clinical trials of glycine recognition site antagonists commence, it will be important to learn if this distinction persists.
Further, the neuroprotective efficacy of glycine recognition site antagonism has recently been demonstrated across a spectrum of both in vitro and in vivo ischemia models. For example, Newell et al. (1995) examined both 7-chlorokynurenic acid and ACEA 1021 in a hippocampal slice culture model subjected to oxygen–glucose deprivation (Newell et al., 1995). Both compounds caused a dose-dependent reduction in CA1 damage. This protection was reversed by increasing the extracellular concentration of glycine. MK-801, a noncompetitive NMDA antagonist, offered similar protection, which was resistant to glycine administration. Others, using in vivo models, have found reduction of focal ischemic infarct volume by i.v. injection of L-687414, a partial agonist of the glycine site, although only a 4-h recovery interval was allowed (Gill et al., 1995). Tsuchida and Bullock (1995) also found protection from glycine recognition site antagonism. In their experiment, rats were subjected to an acute compression injury caused by induction of a subdural hematoma (Tsuchida and Bullock, 1995). ACEA 1021 was found to result in a reduction of infarct size by up to 39%. None of the above in vivo experiments allowed recovery from anesthesia and all used halothane. Halothane itself has previously been shown to offer substantial protection from focal ischemic injury (Warner et al., 1995a). Therefore, the contribution of halothane to the observed protection, either by direct mechanisms or via an interaction with the glycine antagonist, is unknown.
Another cause for concern regarding the reported neuroprotection afforded by glycine site antagonists relates to brain temperature. In the gerbil forebrain ischemia model, i.p. injection of 7-Cl-thio-kynurenate caused a near total protection against CA1 injury, but this protection was substantially attenuated when the effect of the drug on rectal temperature had been accounted for (Pellegrini-Giampietro et al., 1994). With respect to focal ischemia, it has been shown that the filament MCAO model often results in a hyperthermic response to the ischemic challenge (Kuluz et al., 1993; Zhao et al., 1994). We have previously identified a substantial neuroprotective effect for the glycine recognition site antagonists ACEA 1021 and ACEA 1031 using the filament MCAO model (Warner et al., 1995b). However, during those experiments, both drugs were observed to inhibit the hyperthermic response seen in untreated animals during the ischemic insult.
Because of the potential halothane anesthesia confound and also because of reports that the neuroprotective effects of other NMDA receptor antagonists are temperature-dependent (Hoffman and Boast, 1995; Memezawa et al., 1995), the current study was performed to address both the halothane and temperature issues. Attempts were made to minimize exposure to halothane, although the necessity of anesthesia for surgical preparation and removal of the filament remained. Further, brain temperature was controlled both during ischemia and for a substantial interval after onset of reperfusion. We chose to implant the radio thermistor in the contralateral hemisphere to avoid confounds from spreading depression, which could have been elicited by the thermistor. Pilot studies were performed on halothane anesthetized animals to assure that contralateral hemispheric temperatures closely tracked periischemic temperatures in the ipsilateral hemisphere, as determined by a pericranial needle thermistor. Finally, advantage was taken of the fact that ACEA 1021 has recently been reformulated as an i.v. solution, thus allowing more precise control of dosage over prolonged intervals of administration.
Of interest, a dose-response effect was not observed despite a substantial difference in plasma ACEA 1021 concentrations (28 versus 56 μg/ml) for the two treatment groups. This may indicate that maximal neuroprotection occurs at a concentration ≤ 28 μg/ml. Linear regression analysis was performed to determined if a relationship existed between individual plasma concentrations of ACEA 1021 and total infarct volume in treated animals. Such a relationship was absent (r2 = 0.01; p = 0.65). These findings are in contrast to those of our previous work (Warner et al., 1995b). A preischemic plasma ACEA 1021 concentration of 41 ± 9 μg/ml (measured 50 min following 10 mg/kg i.p.) resulted in a 48% reduction in cortical infarct volume when compared to control values. A further 23% reduction of infarct volume occurred at an ACEA 1021 plasma concentration of 69 ± 27 μg/ml (measured 50 min following 30 mg/kg i.p.). We speculate that the greater reduction in infarct volume observed at the higher ACEA 1021 concentrations in that study are attributable to dose-dependent effects on brain temperature. Nevertheless, larger doses of ACEA 1021 could be examined with temperature regulation to assure that the ceiling effect has been achieved in the current study. However, prior work demonstrated that ACEA 1021 concentrations of 84 μg/ml cause loss of righting reflex and absence of withdrawal from deep noxious stimuli (McFarlane et al., 1995). Therefore, doses substantially larger than those administered in the current experiment would likely result in profound sedation.
In conclusion, an NMDA receptor glycine recognition site antagonist was administered i.v. after onset of 75 min of temporary MCAO, and administration was continued for 24 h after reperfusion. This resulted in an ∼60% reduction in mean cortical infarct volume as assessed 7 days later. This occurred despite strict regulation of brain temperature during ischemia and for the first 6 h of recovery. These data are in agreement with those of other studies using more acute models of injury that have identified neuroprotection from this class of compounds and, therefore, confirm an important role for glycine in the pathogenetic mechanism of ischemic brain damage. Further study is required to determine if this class of compounds provides lasting benefit when administration begins at substantial intervals after reperfusion.
Footnotes
Acknowledgment:
This work was supported by USPHS grant RO1 GM39771 and a grant from CoCensys, Inc., Irvine, CA, U.S.A. The authors are grateful to Minthan Tran, Ph.D. for analysis of plasma ACEA 1021 concentrations.