
Abstract
Treatment of brain microvessels with the three endothelin (ET) isoforms resulted in an increase of phosphoinositide turnover by activation of phospholipase C in a dose- and time-dependent manner. Both ET-1 and ET-2 are maximally effective, whereas the effect evoked by ET-3 was smaller. Concomitantly, there was an enhanced production of a platelet-activating factor (PAF)-like material. This was identified by standard and biological probes in platelets, such as induction of aggregation, phosphatidic acid (PA) production, increase of endogenous protein phosphorylation, and reversal of these responses by a PAF antagonist. The effects evoked by endothelins on phosphoinositide metabolism and PAF production were, to a certain extent, dependent on the presence of extracellular Ca2+. In addition, ET induced changes in Ca2+ dynamics, evoking an initial and rapid intracellular mobilization and influx of Ca2+ and, later, a maintained Ca2+ influx. These findings contribute to the understanding of the pathophysiological role of ET in the blood–brain barrier (BBB).
Endothelin (ET), a 21 amino acid peptide, is present in at least three isoforms in the human genome. ET-1 was first isolated from cultured porcine aortic endothelial cells (Yanagisawa et al., 1988), while the existence of ET-2 and ET-3 was predicted following the isolation of genes related to ET-1 (Yanagisawa and Masaki, 1989a). It has been reported that cerebral microvessel endothelium possibly regulates local blood flow within the brain through production of ET (Yoshimoto et al., 1990). Recently, Stanimirovic et al. (1993) reported that ET induces an increase of cerebrovascular endothelium permeability, and McCarron et al. (1993) described the induction by ET of adhesion molecule expression in brain microvascular endothelial cells. On the other hand, endothelial cells from rat brain microvessels express receptor sites for ET. Some of these (receptor sites) are coupled to phospholipase C. Thus, it has been reported that ET-1 stimulates DNA synthesis involving phosphatidylinositol hydrolysis (Vigne et al., 1990) and increases intracellular calcium mobilization in brain capillary endothelial cells (Vigne et al., 1990, 1991). In addition, endothelins inhibit cholera toxin-stimulated adenylate cyclase and increase phospholipase C activity (Ladoux and Frelin, 1991; Stanimirovic et al., 1993; 1994; Purkiss et al., 1994). Although important information may be obtained from analysis of cell cultures, study of intact microvessels can provide useful information to clarify the complexity that exists in vivo in the blood–brain barrier (BBB). In fact, accumulating evidence suggests that the endothelial cells forming the cerebral microvasculature consist of a heterogeneous, rather than homogeneous, cell population, and cultured cell properties may be altered during long-term culture (Kobayashi et al., 1994; Tontsch and Bauer, 1991). Therefore, studies performed on microvessels are important to the understanding of how endothelins are able to regulate the BBB. In this regard, Dallaire et al. (1992) reported that endothelins alter phosphate uptake by isolated capillaries. However, the mechanism by which such actions are evoked is not known.
On the other hand, platelet-activating factor (PAF) is a mediator generated from membrane phospholipids with potent platelet activity and chemotacticproperties (Braquet et al., 1987). PAF production has been demonstrated in endothelial cells (Camussi et al., 1990; Stewart et al., 1990; Whatley et al., 1990, 1992; Clay et al., 1991). Recent evidence suggests that PAF may mediate certain actions of ET-1. Indeed, PAF receptor antagonists are reported to attenuate the bronchoconstrictor action of ET-1 (Filep, 1992), inhibit ET-1-induced increase in intracellular calcium levels (Takayasu et al., 1989), protect against ET-1-induced sudden death in mice and rats (Terashita et al., 1989), and reduce some vascular responses of ET-1 (Takayasu-Okishio et al., 1990; Filep et al., 1991; Kurose et al., 1991). Furthermore, ET-1 has been shown to stimulate PAF synthesis in mesangial cells (López-Farré et al., 1991; Montera et al., 1993).
Taking into account all of these considerations, this study was performed on intact cerebral microvessels mainly to examine the action of the endothelins on phosphoinositide metabolism. In addition, the possibility that PAF release arises by ET action was also examined.
METHODS
Chemicals
Carrier-free [32P]orthophosphate, [3H]myo-inositol (specific activity of 14.3 Ci/mmol), [3H]sodium acetate (specific activity 3.6 Ci/mmol), and 45CaCl2 (specific activity 13.4 mCi/mg) were purchased from Amersham International. Endothelins and quinacrine were from Sigma Chemical Co, (St Louis, MO, U.S.A.). Fura-2 AM was from Molecular Probes Inc. (Eugene, OR, U.S.A.). High performance thin layer chromatography plates were purchased from Merck (Darmstad, Germany). Diacylglycerol (DAG) assay system was obtained from Amersham. All other reagents were of the highest analytical grade available.
Isolation of brain microvessels
Cerebral microvessels were isolated basically as described previously (Catalán et al., 1989b). Brains were removed from male Wistar rats by decapitation and placed in a cold medium (buffer A) containing 15 mM HEPES (pH 7.4), 103 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, and 10 mM glucose. Brainstem, cerebellum, and meninges were carefully removed and discarded, and the cerebral cortical grey matter was homogenized using a hand-driven pestle (20 strokes at 400 rpm) in 5 vol of buffer A. After centrifugation at 1,000 × g for 10 min at 4°C, the pellet was resuspended in 4 vol of buffer A containing 15% dextran and centrifuged at 5,000 × g for 10 min at 4°C to remove the myelin. The resulting pellet, consisting mainly of microvessels and erythrocytes, was resuspended in buffer A and poured onto a nylon sieve, pore-size 150 μm. The filtrate was subsequently passed over a column (2.5 × 8 cm) of 0.45 mm diameter glass beads. Microvessels retained on the glass beads were separated by decantation and centrifugation at 5,000 × g for 10 min at 4°C. Finally, the pellet was resuspended at a final concentration of 0.1–0.5 mg protein/ml in buffer A supplemented with 1 mg/ml bovine serum albumin, 1 mM CaCl2, 1 mM inositol, and 1 mM cytidine (buffer B). Before assays, microvessels were preincubated for 15 min at 37°C under an atmosphere of 95% O2 and 5% CO2 (vol/vol). The purity of the preparation was quite satisfactory from both morphological and biochemical criteria, and neuronal contaminants were negligible.
Assays of phosphoinositide turnover
To analyze the effect of endothelins on 32P incorporation into phospholipids, microvessels were prelabeled in a phosphate-free medium in the presence of 100 μCi/ml [32P]orthophosphate for 30 min. Thereafter, ET treatment was carried out for the appropriate times. In some experiments, the effects of endothelins in the presence or absence of 250 μM quinacrine or 5 mM EGTA were also analyzed. Incubations were stopped by removing the medium and replacing it with cold chloroform/methanol/13 N HCl (20:100:0.75, vol/vol/vol) as described previously (Catalán et al., 1991). Then, phospholipids were extracted according to Jolies et al. (1979). Phospholipid separation was performed by thin layer chromatography (TLC) using silica gel high performance TLC plates impregnated with 1% potassium oxalate in methanol/water (2:3, vol/vol), and chromatograms were developed as described previously (Catalán et al., 1991). Dried plates were autoradio-graphed overnight using AGFA x-ray film. Silica gel sections corresponding to labeled phosphoinositides, phosphatide acid (PA), and PAF were scraped off into vials and the radioactivity determined.
DAG content was determined essentially as described by Preiss et al. (1986), with a bacterial DAG kinase to convert cellular DAG to 32P-labeled PA, using the DAG assay system (Amersham International). Samples were extracted with chloroform/methanol, and 32P-PA in the chloroform extract was quantified by liquid scintillation, after separation by TLC as described above.
To analyze the production of inositol phosphates by treatment with endothelins, microvessels were incubated in an inositol-free medium at 37°C with 20 μCi/ml [3H]myo-inositol. After 120 min, 10 mM LiCl was added and ET treatment was carried out. Incubations were stopped by addition of 1 vol of 20% cold trichloroacetic acid, and extracts were washed five times with water-saturated ether and then neutralized before analysis. Inositol phosphates were separated by anion-exchange high-performance liquid chromatography (HPLC) with Ultrasil-SAX column (4.6 × 250 mm; Beckman) as described previously (Batty et al., 1985).
Measurement of calcium movement
To estimate Ca2+ efflux, cerebral microvessels, resuspended in the buffer A described above, were equilibrated with 1 mM 45CaCl2 (1.5 μCi/ml) at 35°C for 45 min. Then, 10−7 M ET was added, and the reaction stopped at different times. Samples were filtered rapidly through Whatman GF/B filters, washed three times with a solution containing 10 mM Tris HCl, pH 7, and 200 mM sucrose. Radioactivity was determined by a standard liquid-scintillation technique.
In order to measure intracellular Ca2+ levels, microvessels were preincubated in buffer B for 15 min and then were loaded with 5 μM fura-2 AM at 37°C for 30 min under an atmosphere of 95% O2 and 5% CO2 (vol/vol). Microvessels were washed to release extracellular dye, and resuspended in Ca2+-free buffer B supplied with 250 μM sulfinpirazone. Samples were incubated for 10 min in the presence of 0.7 mM Ca2+ in a final volume of 0.5 ml. Finally, microvessels were resuspended in the same buffer B with or without Ca2+ in a cuvette with constant shaking at 37°C before addition of ET. Calcium measurements were determined in a volume of 2.5 ml by fluorescence using a RS1000 Schoeffel (Schoeffel Instruments, Ltd.) spectrophotofluorimeter. Excitation wavelength was 340 nm and emission was monitored at 505 nm. Fluorescence measurements were calculated as described previously (Grynkiewicz et al., 1985) after subtracting background fluorescence measured in microvessels not loaded with fura-2. Maximum bioavailability, Fmax, was determined after lysing microvessels with 160 μM digitonin in the presence of 1.5 mM Ca2+, and Fmin was measured after adding 10 mM EGTA and 20 mM Iris, pH 8.7. Higher concentrations of EGTA produced no further decrease in Fmin. The apparent disassociation constant, Kd, of the fura-2/Ca2+ complex was 224 nM.
Production and biological activity of PAF
After phospholipid separation described above, the spot corresponding to PAF was scraped off the plates, and extracted with chloroform/methanol/water (1:2:0.8, vol/vol/vol) as described previously (Hanahan, 1990). Samples were dried under nitrogen and resuspended in Tyrode-HEPES, pH 7.4, containing 134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM NaH2PO4, 1 mM MgCl2, 5 mM HEPES, 5 mM glucose, and 0.3% bovine serum albumin. To analyze the biological activity of PAF, washed rabbit platelets, obtained as described previously (Catalán et al., 1993a), were treated with the PAF fraction isolated as described above. Incubations were performed as previously reported (Catalán et al., 1993a). The PAF activity of the lipid extracted by this fashion was determined by testing both PA increase and protein phosphorylation from washed rabbit platelets, which have been previously loaded with [32P]orthophosphate (Catalán et al., 1989a).
In some experiments incorporation of radioactive precursors into PAF and its modulation by ET was studied. For this, isolated microvessels were incubated with either 100 μCi/ml [32P]orthophosphate or 200 μCi/ml [3H]acetate for 30 min or 15 min respectively. Then, 10−7 M ET-1 was added. After different times (from 1 min to 10 min), radioactivity incorporation into PAF was measured.
Analysis of results
Each result represents the mean of triplicate determination ± SD. Statistical analysis of the data was done using Student's t-test. Differences with a p-value of <0.05 were considered statistically significant.
RESULTS
Effects of ETs on phosphoinositide turnover
Treatment of brain microvessels prelabeled with [32P]orthophosphate with the three ET isoforms resulted in an increase of phosphoinositide turnover in a dose-dependent manner, 10−7 M being the maximal effective dose used. Figure 1 shows the dose-response curves of ET on 32P incorporation into phosphatidylinositol 4,5-bisphosphate (PIP2) and PA. Phosphatidylinositol 4-monophosphate (PIP) and phosphatidylinositol (PI) showed the same pattern of variation by ETs (data not shown). It is noteworthy that the effect of ET-3 was smaller than that of other ET isopeptides. The EC50 of both ET-1 and ET-2 was 10−8 M, whereas ET-3 showed an EC50 of 5 × 10−8 M. On the other hand, 1(T7M of any ET isoform increased PA formation at 1 min, peaked at 5 min, and then was decreased at 15 min (Fig. 2). In addition, ETs caused an early decrease of 32P incorporation into PIP2 at 1 min, then returned to basal through 15 min. Effects of both ET-1 and ET-2 were again more pronounced than that of ET-3. These effects were, at least in part, calcium dependent. Thus, incubation of microvessels in a Ca2+-free medium with 5 mM EGT A resulted in a reduction of the 1 min ET-1 induced variations by >50%, abolishing the ET action at 5 min of treatment (Fig. 3).
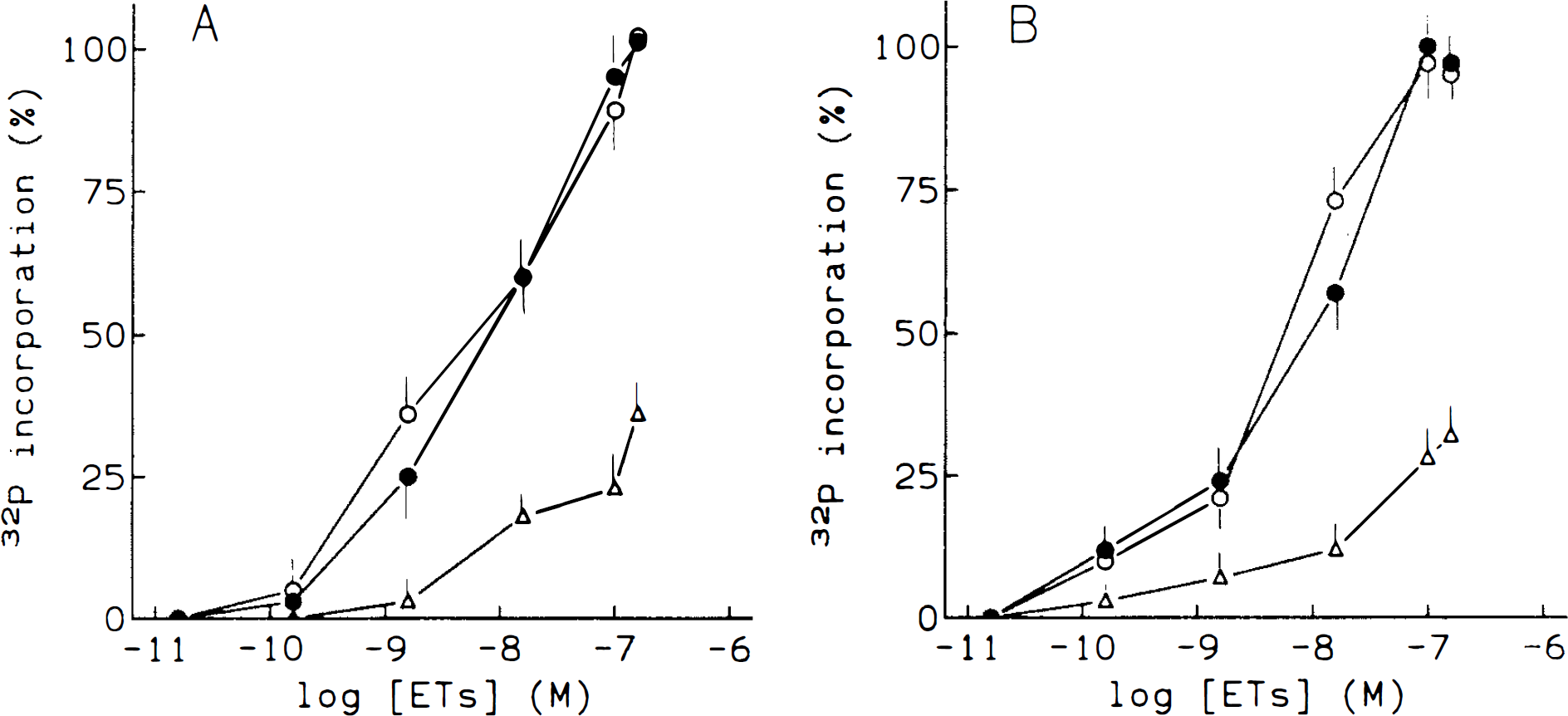
Dose-response relationship between ET concentration and PIP2 and PA 32P-labeling. After prelabeling for 30 min with 100 μCi/ml [32P]-orthophosphate, microvessels were treated with different concentrations of ET. The time of treatment was 1 min for PIP2 determination (
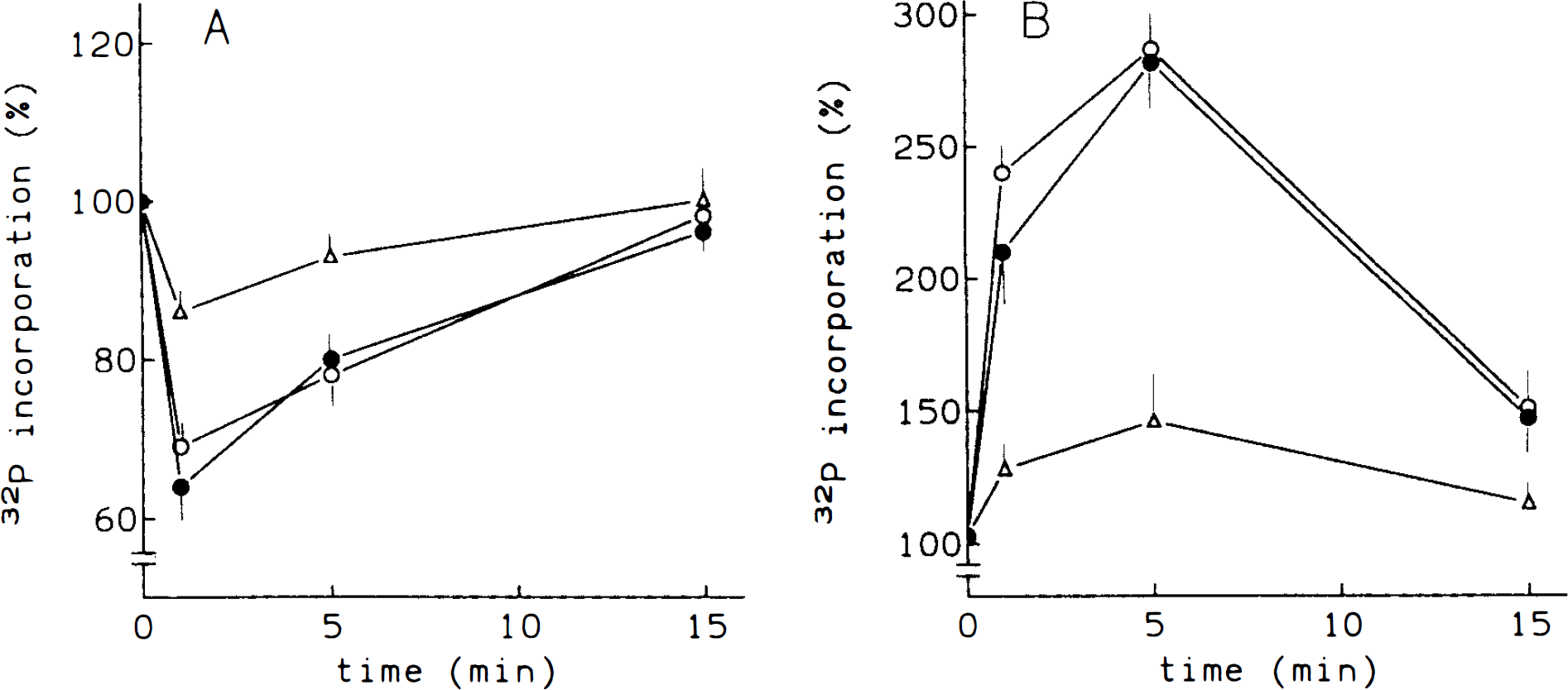
Time-course of ET effects on [32P]-orthophosphate incorporation into PIP2 (
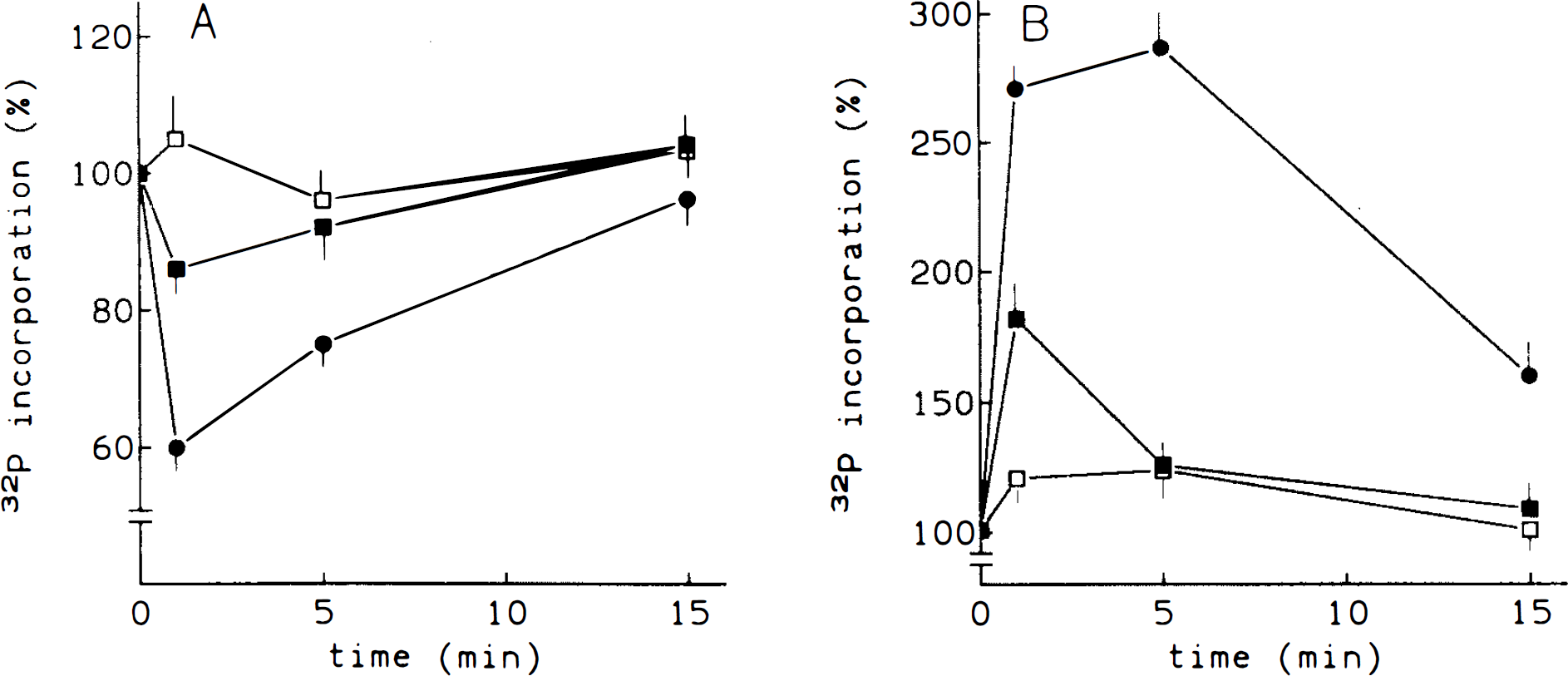
Role of extracellular Ca2+ in the ET effect on phosphoinositide turnover. Microvessels were prelabeled as in the legend to Fig. 1 and treated with 10−7 M ET-1. In some incubations, 5 min before ET treatment, EGTA was added (final concentration was 5 mM). The radioactivity into PIP2 (
The measurement of phosphoinositide metabolites (i.e., DAG and inositol phosphates) showed that the variations elicited by ET are the result of phospholipase C activation (Figs. 4 and 5). Thus, the addition of 10 −7 M ET-1 to brain microvessels induced a time-dependent increase of DAG levels (Fig. 4). Accumulation of DAG was detected as early as 30 s, and then DAG levels decreased and were maintained for at least 15 min.
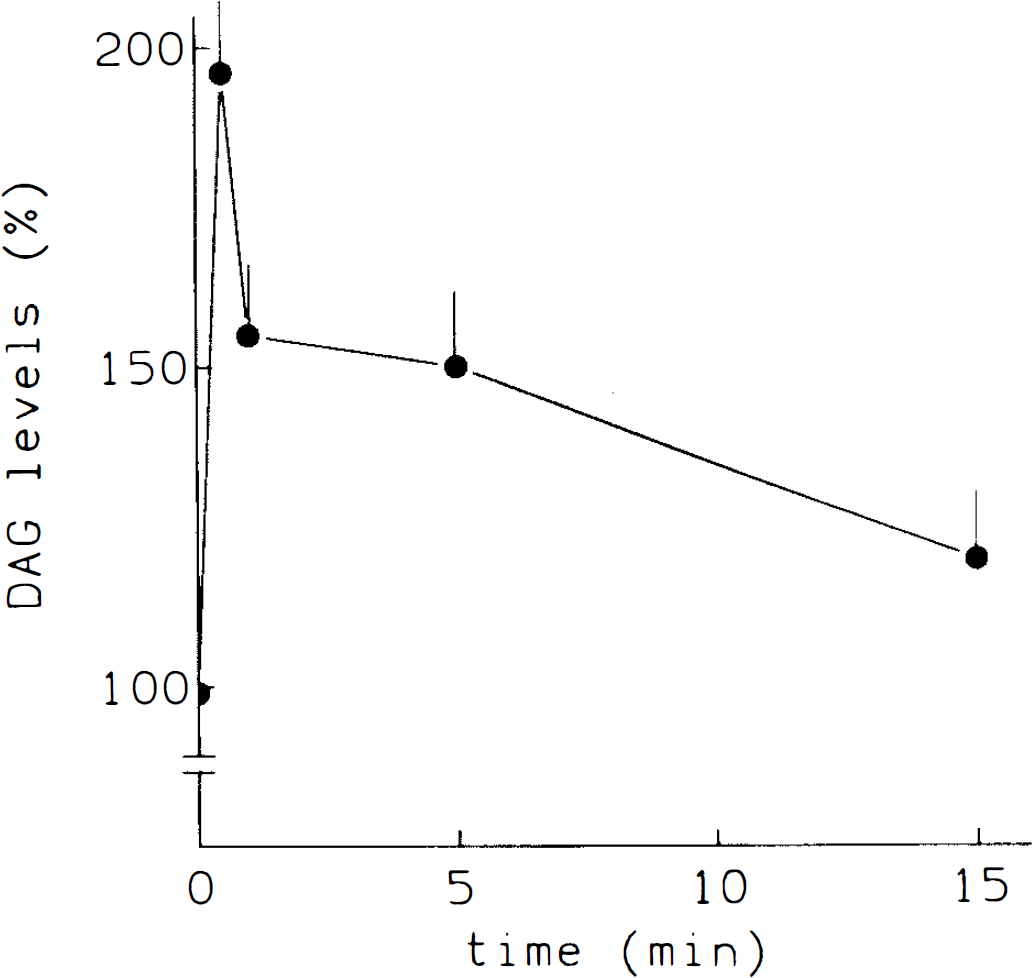
Effect of ET-1 on DAG production. Microvessels were treated with 10−7 M ET-1 for the times indicated. Afterwards, DAG was extracted and quantified as described in Methods. Results are expressed as percentage of variation with respect to control values at zero time (100). Basal value: 0.7 nmol/mg protein. Bars represent standard deviation, n = 3. All values were statistically significant (p < 0.05).
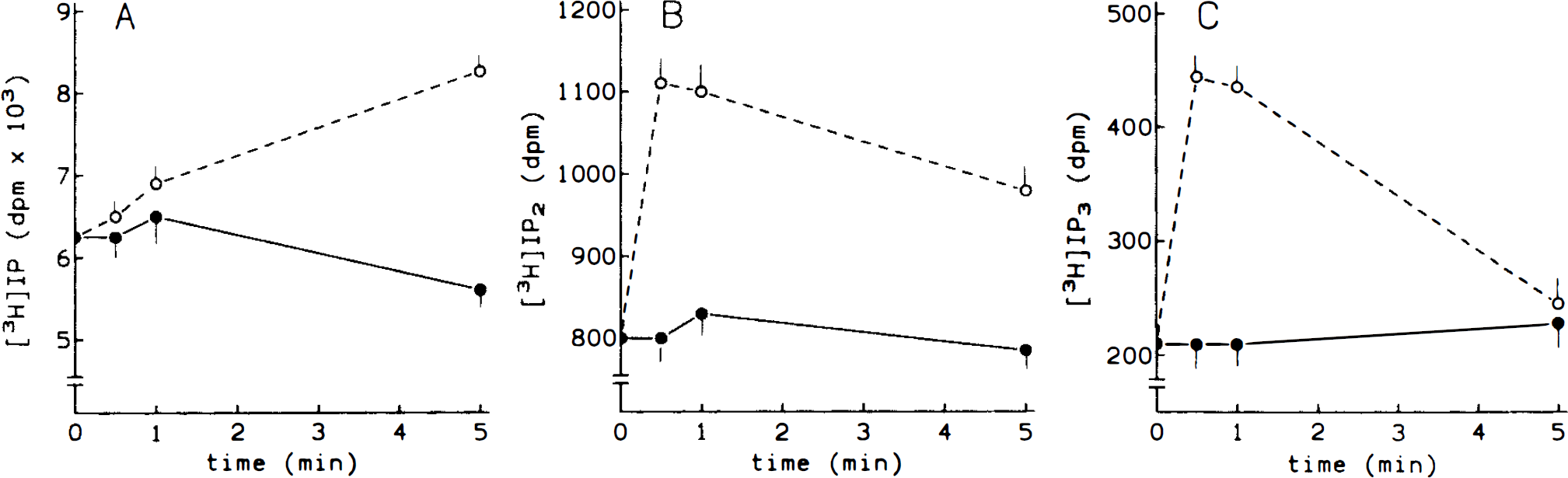
ET-1-induced inositol phosphates production. Cerebral microvessels were prelabeled with [3H]myoinositol (20 μCi/ml) for 120 min and then 10−7 M ET-1 was added for the times indicated. Results are expressed as dpm incorporated into each inositol phosphate. A, IP; B, IP2; C, IP3. • control; ˆ, ET-1. Basal values: 2 pmol/mg protein, IP; 0.25 pmol/mg protein, IP2; 0.08 pmol/mg protein, IP3. Bars represent standard deviation, n = 3. All values (except for IP at 0.5–1 min and for IP3 at 5 min) were statistically significant (p < 0.05).
The time course for accumulation of inositol phosphates in response to ET-1 was also determined (Fig. 5). Both inositol 1,4,5-trisphosphate (IP3) and inositol 1,4-bisphosphate (IP2) levels rose rapidly at 30 s. Inositol 1-monophosphate (IP) levels increased linearly due to the inclusion of Li+ during the stimulation period, which inhibits the hydrolysis of IP2 and mainly that of IP.
Involvement of Ca2+ in ET effect
To test the effect of ET-1 on intracellular Ca2+, experiments were performed using fura-2-loaded microvessels. In the presence of extracellular Ca2+, 10−7 M ET-1 induced an initial and rapid Ca2+ peak followed by a second sustained phase (Fig. 6A). When ET-1 was added to microvessels incubated in a Ca2+-free medium, the transient increase was attenuated and the sustained phase abolished (Fig. 6B).
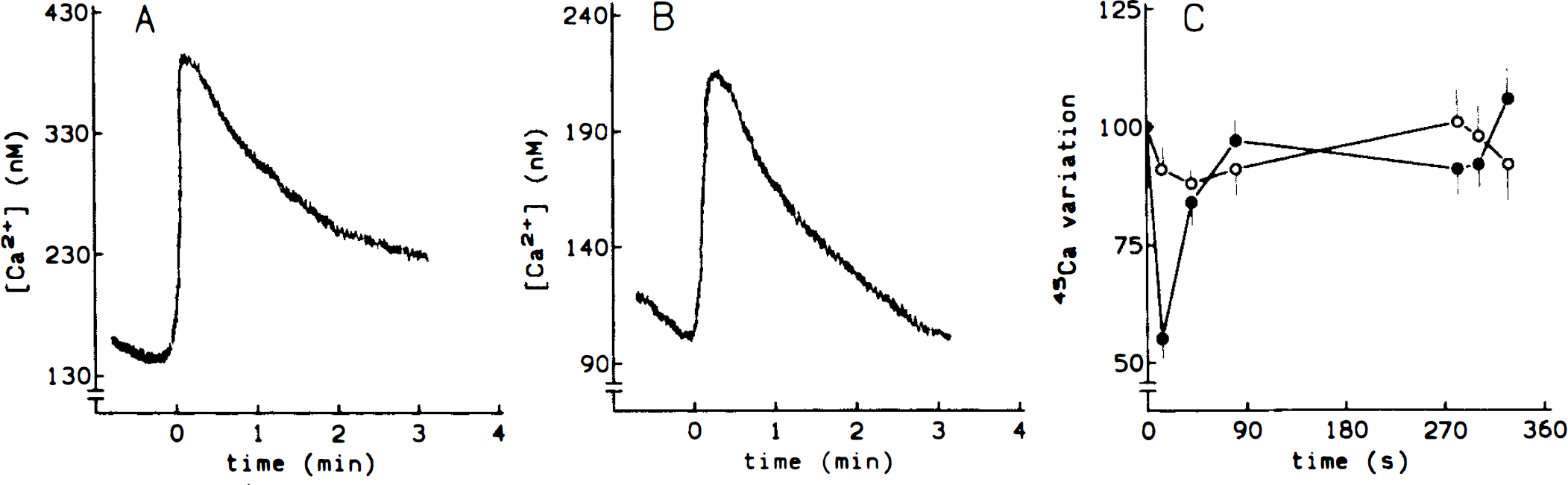
Effects of ET-1 on Ca2+ movements. Effect of ET-1 on Ca2+ intracellular levels (
On the other hand, results obtained when microvessels were equilibrated with 45Ca2+ for 45 min to label the internal stores, showed that 10−7 M ET causes a rapid, but transient, increase in the Ca2+ efflux (Fig. 6C). Thus, after 45 s, 45Ca2+ reached basal levels. Untreated microvessels did not show any oscillations of radioactivity.
PAF production by ET
After ET-1 treatment, 32P-prelabeled brain microvessels were able to produce 32P-PAF. Once phospholipids were fractionated, PAF was located on the TLC plate between the area corresponding to sphingomyelin (Rf value = 0.28) and lyso phosphatidylcholine (Rf value = 0.14) as described previously (Hanahan, 1990). The area comigrating with authentic PAF was scraped off the plates, extracted, and its biological activity analyzed.
The extracted PAF was added to washed rabbit platelets, and aggregation rapidly occurred (data not shown). Furthermore, ET-induced PAF also caused PA formation and 40 kDa protein phosphorylation (Fig. 7, panels A and B respectively). Pretreatment of platelets with 10 μM WEB 2086, a specific PAF receptor antagonist, reduced all these effects evoked by ET-induced PAF in platelets (Fig. 7).
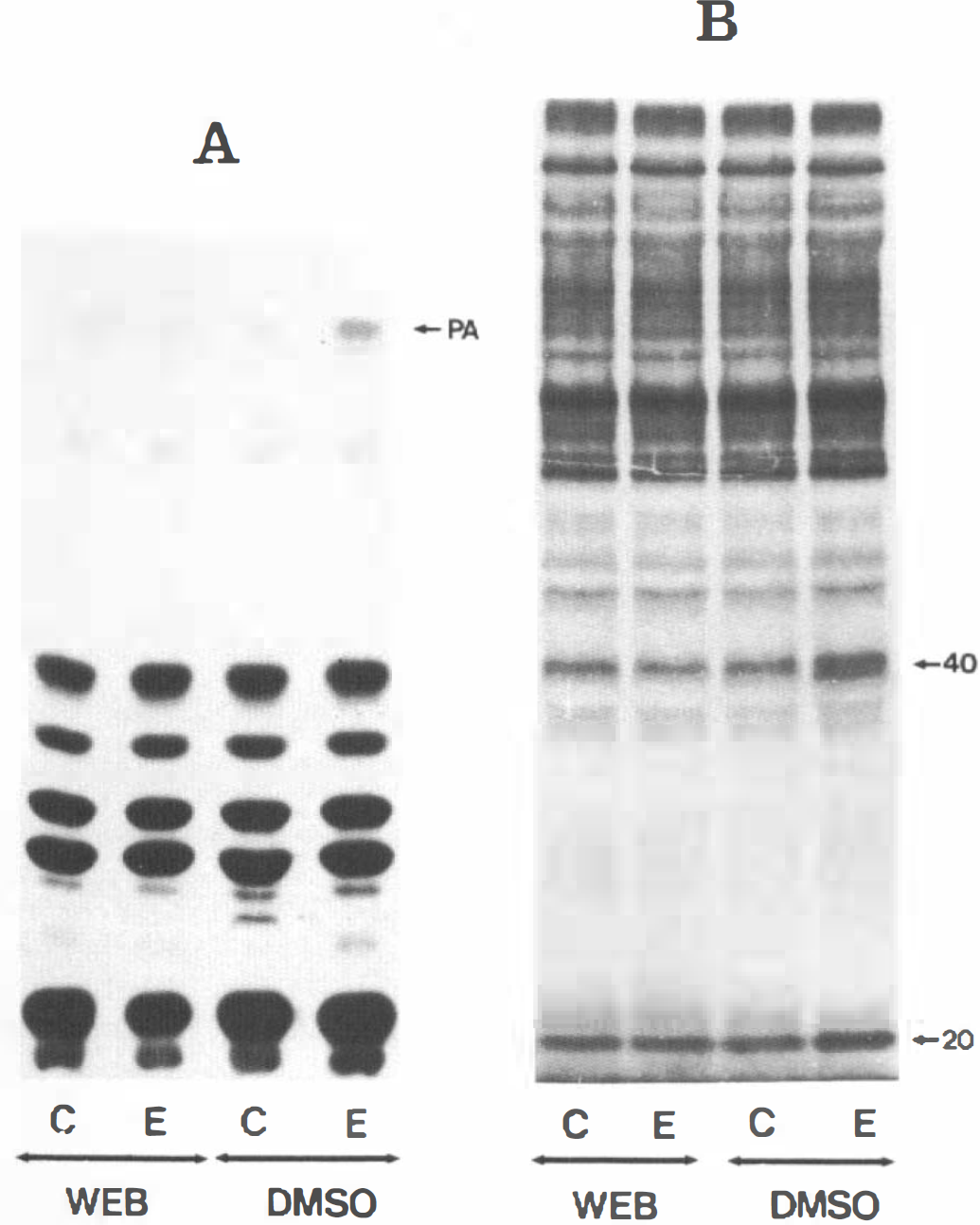
Platelet responses to PAF-like material produced by ET stimulation. Rabbit platelets (108 cells/ml) were labeled with 100 μCi/ml [32P] orthophosphate for 1 h. Then, they were stimulated with the PAF-like material extracted from lipids of ET-stimulated microvessels (E) or with those eluted from the corresponding chromatographic area from the plate after lipid separation of untreated microvessels (C) in the presence or absence (DMSO) of 10 μM WEB 2086, a PAF antagonist. Incubations were stopped and phospholipids or endogenous phosphoproteins were analyzed, as described under Methods. Panel
To test whether PAF was released to the extracellular medium, some incubations were finished by centrifugation, and aliquots of the supernatant and resuspended sediment were measured for radioactivity. Results showed that PAF was not released to the extracellular medium (data not shown).
Using 32P as labeled precursor of PAF, incorporation of label into PAF after treatment with 10−7 M ETs was studied. As can be seen in Fig. 8A, maximal incorporation was observed after 5 min of treatment with both ET-1 and ET-2. After 15 min, incorporation returned to basal value. Figure 8 also shows the dose-response curves for the stimulation of PAF production. Maximal stimulation was evoked by both ET-1 and ET-2 (10−7 M). In any case, no effect was observed when ET-3 was assayed (Fig. 8B).
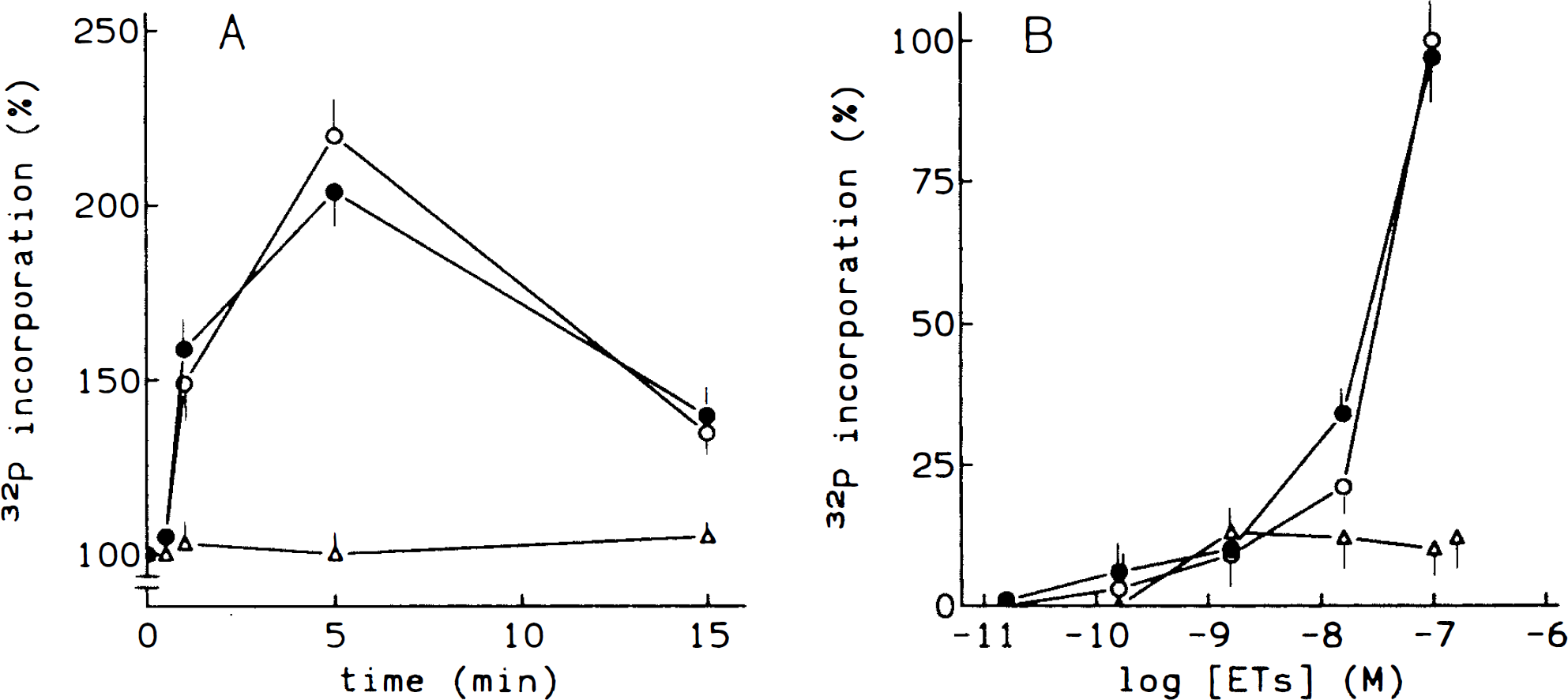
Effect of endothelins on [32P]-PAF production. Time course of ET-stimulated PAF production.
Additional experiments were carried out to further characterize the ET-evoked PAF production. Thus, it is noteworthy that ET-1 did not stimulate 32P incorporation into PAF in the presence of either 5 mM EGTA or 250 μM quinacrine, an inhibitor of phospholipase A2 (Fig. 9).
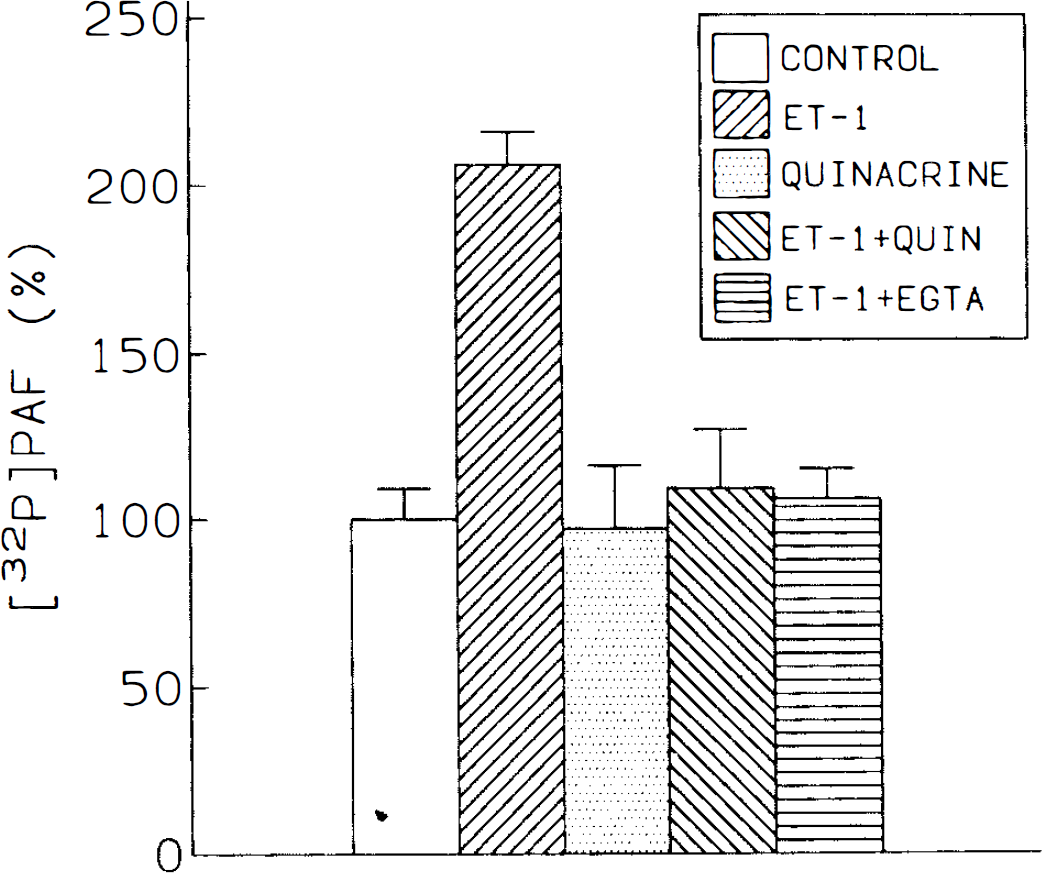
Involvement of Ca2+ and phospholipase A, activity in PAF production by ET. Prelabeling with [3P]-orthophosphate was carried out as indicated in the legend to Fig. 1. Then, microvessels were treated for 2 min with 5 mM EGTA or for 10 min with 250 μM quinacrine. Then, 10−7 M ET-1 was added and, after 5 min, PAF was extracted and analyzed as in Methods. Results are expressed as percentage of control value (100). Bars represent standard deviation, n = 3. Values for ET-1 were statistically significant (p < 0.05).
DISCUSSION
Several important points emerge from the present data. Most of all, ETs are effective in inducing phosphoinositide hydrolysis and subsequent resyn-thesis in intact cerebral microvessels. This hydrolysis is mediated by phospholipase C activation, as shown by the rapid production of inositol phosphates together with an increase of DAG mass and PA labeling. However, we must take into consideration that other phospholipases (i.e., phospholipase D) and/or other phospholipids (i.e., phosphatidylcholine) might be involved in overall PA production as well as DAG generation.
There is previous evidence that many ET responses are mediated by phospholipase C activation in other tissues and cells (see review by Yanagisawa and Masaki, 1989b). However, there appear to be substantial differences in the mechanism of action of ET among different species and cells. Therefore, the study and the elucidation in the BBB of the signal transduction pathways involved in ET actions are relevant in determining the significance of this peptide in the BBB. Intact cerebral microvessels constitute a tissue for which no previous studies on ET signal transduction have been reported. Studies done in isolated capillary endothelial cells (Vigne et al., 1990, 1991; Ladoux and Freiin, 1991; Stanimirovic et al., 1993, 1994; Purkiss et al., 1994) show that both phospholipases C and A2 are involved. Since cerebral endothelial cells rapidly lose their characteristics of differentiated barrier in culture (Kobayashi et al., 1994; Tontsch and Bauer, 1991; Risau and Walburg, 1990), intact cerebral microvessels appear to be a better model for the study of BBB functionality than are cultured endothelial cells. In fact, there are controversial data about the ability of these cells in culture to secrete ET (Yoshimoto et al., 1990; Vigne et al., 1990) and about the kinetic characteristics of inositol phosphate production by stimulation with the peptide (Vigne et al., 1990; Ladoux et al., 1991; Stanimirovic et al., 1993, 1994; Purkiss et al., 1994). Furthermore, it has been described that cultured endothelial cells could be a kind of injured cell since they produce more ET than does the microvessel itself (Yoshimoto et al., 1990).
It is noteworthy that the potency of both ET-1 and ET-2 to stimulate phospholipase C in intact microvessels was higher than that of ET-3, in accordance with data reported for cerebrovascular endothelial cells (Stanimirovic et al., 1993, 1994; Purkiss et al., 1994). Actually, ET receptors in endothelial cells from brain microvessels are coupled to phospholipase C, but recognize ET-3 with low affinity, in contrast to the selectivity shown for ET-1 (Vigne et al., 1991). This fact could indicate that they are of the ETA type, since ETB receptors show equipotent affinity for all three ETs, according to the classification of ET receptors (Sakurai et al., 1992). In agreement with this fact, the ETA receptor antagonist BQ 123 has been shown to inhibit the IP3 formation elicited by ET in endothelial cells from human microvessels (Stanimirovic et al., 1994).
Besides stimulating phosphoinositide turnover, both ET-1 and ET-2, but not ET-3, treatments evoked the appearance of 32P radioactivity in a chromatographic band when phospholipids were fractionated. Identification of this band by standard and biological probes demonstrated that it was a PAF-like material. Evocation of aggregation, production of PA, and increase of endogenous protein phosphorylation in platelets, together with the reversal of these responses by a PAF antagonist, were used as biological probes. The finding that ET evokes PAF synthesis agrees with previous observations (López-Farré et al., 1991; Montero et al., 1993) and also confirms other studies suggesting that PAF mediates certain actions of ET (Filep et al., 1991; Filep, 1992; Kurose et al., 1991; Takayasu et al., 1989; Takayasu-Okishio et al., 1990; Terashita et al., 1989).
To our knowledge, this is the first report of PAF production by cerebral microvessels. Our present results show that the characteristics of PAF production by cerebral microvessels are similar to those of PAF release by endothelial cells from other vessels. PAF is generated in these cells after agonist activation and requires the presence of extracellular cations and the activation of phospholipase A2, indicating that the remodeling pathway of PAF biosynthesis is involved (Camussi et al., 1986; Whatley et al., 1990). In addition, none of the endothelial PAF is released into the extracellular medium (Stewart et al., 1990), a fact that has led to the proposal that PAF is expressed in the outer leaflet of the endothelial cell membrane, causing contacting neutrophils to adhere. In contrast, the PAF produced by mammalian central nervous system neurons is detected in the cells and in the extracellular medium. In addition, its production is practically independent of calcium, a fact that could suggest that the de novo biosynthesis pathway is the main route for PAF production (Yue et al., 1990). Therefore, from the data reported here, it is quite logical to assume that PAF from cerebral microvessels is mainly, if not exclusively, produced by endothelial cells.
We have previously reported that in cerebral microvessels, translocation of protein kinase C is evoked by PAF (Catalán et al., 1993b). Therefore, we suggest that PAF might have a second messenger role in ET action on cerebral microvessels, as has been suggested in other biological systems (Stewart et al., 1990).
Previous studies, performed with different cell types, suggest that the effects of ET are generally mediated by inositol phospholipid turnover, and require extracellular Ca2+ (Yanagisawa and Masaki, 1989b; Highsmith et al., 1992). Results of our experiments performed with cerebral microvessels in Ca2+-free media, showing that ET actions are impaired in these conditions, agree with results from other tissues. However, the earliest ET-induced (1 min) phosphoinositide turnover is only partially Ca2+-dependent in contrast to the fully Ca2+-dependent responses observed when the time of ET treatment was ≧5 min (i.e., phosphoinositide turnover and PAF production). With respect to ET-induced changes in Ca2+ dynamics in cerebral microvessels, the ability of the peptide to induce an initial Ca2+ spike, followed by a prolonged plateau, suggests the involvement of different Ca2+ signaling pathways. Results indicated that the observed initial spike is likely to be due to both an IP3-evoked mobilization of intracellular Ca2+ and a Ca2+ influx. On the other hand, since the plateau is abolished by extracellular Ca2+ depletion, we suggest that this is due to a Ca2+ influx. Similar Ca2+ movements induced by ET have been previously reported in other biological systems (see review by Highsmith et al., 1992).
The Ca2+ influx might be due to channels gated by IP3/inositol tetrakisphosphate; channels gated by other second messengers, such as DAG (Berridge and Irvine, 1989); or to voltage-dependent channels. Previous data on the type of Ca2+ membrane channels involved in ET action have demonstrated that both voltage-sensitive and insensitive channels are implicated in a cell-specific fashion (see review by Highsmith et al., 1992). In endothelium from cerebral microvessels, partial involvement of voltage-dependent Ca2+ channels in ET-induced IP3 formation, arachidonate release, and the increased permeability has been reported (Stanimirovic et al., 1993). In addition, we have observed that ET evokes a rapid Ca2+ efflux in cerebral microvessels. In agreement with this phenomenon, we must point out that the peptide has been shown to evoke Ca2+ efflux in vascular smooth muscle due to mobilization of intracellular Ca2+ stores (Bialecki et al., 1989).
Involvement of ETs in the pathogenesis of vascular disorders, such as stroke, hypertension, and subarachnoid hemorrhage (Sakurai and Goto, 1993), and in the function of the BBB (Vigne et al., 1991) has been reported. Therefore, a precise knowledge of the molecular mechanism of ET action on BBB microvessels is a requisite to the development of an effective therapy. As an example, from the evidence reported here, the therapeutic use of both PAF and Ca2+ antagonists in BBB diseases in which ETs are involved has the potential for effectiveness.
Footnotes
Abbreviations used
Acknowledgment
The authors are indebted to Miss M. Sanz and Mr. J. Palaci'n for their valuable assistance. This work was supported, in part, by DGICYT, FIS, CAM and Fundación Ramón Areces.