
Abstract
We developed an antibody specific to β-amyloid precursor protein (βAPP) fragments possessing the exact amino terminus of the β-amyloid peptide and examined its induction in postischemic hippocampus. In control hippocampus, this APP fragment was lightly observed in pyramidal neurons of CA sectors and dentate granule cells. Transient forebrain ischemia enhanced accumulation of the APP fragment in CA1 pyramidal neurons. Seven days after the ischemia, while the APP fragment was still observed in dentate granule cells and CA3 neurons, it disappeared in dead CA1 neurons. While astrocytes did not show in any immunoreactivity throughout the experiment, those in the CA1 sector showed moderate immunoreactivity 7 days after the ischemia. The APP fragment has a cytotoxic effect on cultured neurons. These results suggest that the accumulation of the cytotoxic APP fragment in CA1 neurons may play a role in the development of delayed neuronal death after the ischemic insult.
Transient global forebrain ischemia in rodents induces selective degeneration affecting hippocampal CA1 and CA4 neurons with relative sparing of CA3 neurons and dentate granule cells. Although the degeneration of CA4 neurons begins within hours after the ischemic insult, CA1 neurons remain intact for up to 2 days and then degenerate (delayed neuronal death) (Kirino, 1982). The molecular mechanisms responsible for differential neuronal vulnerability to ischemic injury are incompletely understood (Schmidt-Kastner and Freund, 1991). In the previous study, we showed that an amyloidogenic process was induced in the early phase of postischemic hippocampus (Saido et al., 1994). Here we examined the detailed distribution of the cytotoxic fragment of the β-amyloid precursor protein (βAPP) in the ischemic hippocampus and the role of its accumulation in development of the delayed neuronal death of CA1 neurons.
The antibody specific for the amino terminus of β-amyloid peptide (Aβ) was produced and its characteristics were described in a previous study (Saido et al., 1994). This antibody (Ab-9204) recognizes both Aβ and the COOH terminal fragment of βAPP consisting of 99 or 100 amino acid residues (APP-C100), but not intact βAPPs (βAPP695, βAPP751, and βAPP770) or the physiologically secreted form of βAPP (Fig. 1).
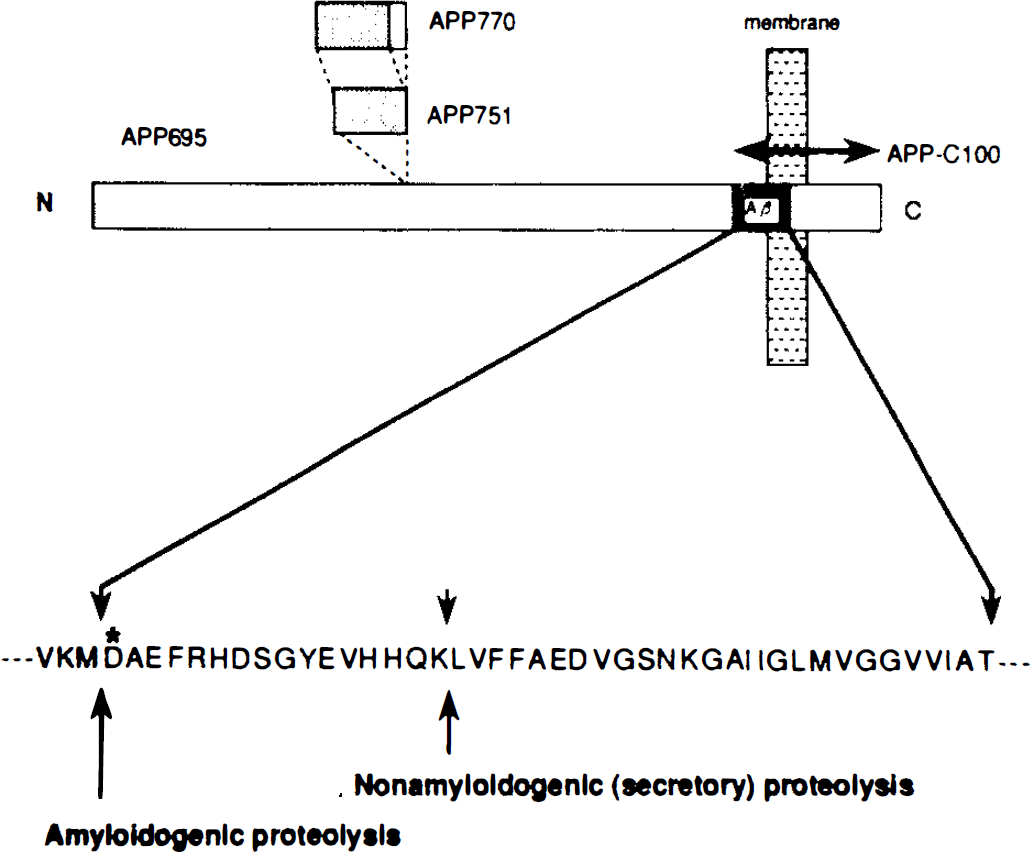
Schematic diagram of the β-amyloid precursor protein (βAPP). Epitope of the novel antibody (Ab-9204) used in the present study is hexamer peptide, DAEFRC (asterisk), in N-terminus of β-amyloid peptide (Aβ). Ab-2904 recognizes Aβ and APP-C100, but not βAPP695, βAPP751, BPP770, or the secreted form of βAPP.
MATERIALS AND METHODS
Adult female Mongolian gerbils weighing 50–70 g (Japan Clea) housed under diurnal lighting conditions and allowed food and water ad libitum were anesthetized during the ischemia by diethyl ether and room air. Body temperature was maintained at 36–37°C with a homeothermic heating blanket (CMA, Stockholm, Sweden) during the anesthesia. Following a midline cervical incision, both common carotid arteries (CCAs) were dissected in 40 gerbils and occluded with small aneurysmal clips. After a 10-min occlusion, the clips were removed to allow recirculation. In 40 sham-operated animals, CCAs were dissected but not occluded. Four animals from each group (ischemic and sham) were killed at intervals of 4 h, 1 day, 2 days, 3 days, and 7 days after recirculation. At each time point, the animals were euthanized with an overdose of sodium pentobarbital (60 mg/kg i.p.) and then perfused at 4°C through the ascending aorta with phosphate-buffered saline (PBS), followed by 2% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 4°C for immunohistochemical study. The perfused brains were removed and fixed with the same fixative for 24 h. Next the brains were immersed in PBS containing 10–20% sucrose for 24 h and processed for immunohistochemistry. To assess tissue architecture, 5-μm sections from each hippocampus were prepared after the fixation and stained for cresyl violet.
Immunohistochemical study was performed essentially as previously described. The brain was coronally sectioned at 20 μm on a cryostat. The sections were incubated for 15 min at room temperature with 0.3% H2O2 in PBS followed by 3% normal goat serum in PBS for 20 min. They were then incubated for 24 h at 4°C with a 1:500 dilution of Ab-9204. The sections were incubated for 2 h in biotinylated anti-rabbit IgG made in goat (Vectastain, ABC kit) and then for 2 h with avidin–biotin–peroxidase complex at room temperature. Immunolabel was visualized with 0.015% diaminobenzidine tetrahydrochloride (DAB) and 0.003% H2O2 in 50 mM Tris-HCl buffer (pH 7.6).
To specify the glial cell types, one of the two serial thin (4-μm) sections of the hippocampus on day 7 was processed for glial fibrillary acidic protein (GFAP) staining as described previously (Yokota et al., 1994), and the other section was processed for APP-C100 staining. For GFAP staining, sections were treated in methanol containing 0.3% hydrogen peroxide. Sections were incubated in 10% normal goat serum followed by incubation in monoclonal mouse anti-human GFAP (Dako) diluted 1:100 for 24 h at room temperature. Then sections were incubated in peroxidase-conjugated goat anti-mouse IgG. Sections were incubated in DAB as described already. Additionally, double-immunolabeling histochemistry for APP-C100 and GFAP was performed to clarify colocalization of the both antigens in astrocytes. At first, sections were immunostained by Ab-9204 and visualized with DAB as described. Then the sections were incubated in monoclonal mouse anti-human GFAP and followed by incubation in peroxidase-conjugated goat anti-mouse IgG. GFAP was visualized by 4-chloro-1-naphthol.
For immunoblot analysis, three sham-treated animals and another three animals treated with 10-min ischemia were perfused with PBS at 1 day after operation. The hippocampus was dissected under a surgical microscope, immersed in liquid nitrogen, and remained frozen at −85°C until further processed. The collected samples were homogenized in the solubilization buffer for sodium dodecyl sulfate Polyacrylamide gel electrophoresis, and after brief sonication and heat treatment, aliquots were subjected to Western blot analysis. Western blotting was performed by using Ab-9204 as described in the previous study (Saido et al., 1992). Each immunolabeled band was densitometrically quantitated by a videodensitometry system (ACI Japan) and expressed as percentage of sham density. To establish the specificity of Ab-9204, an immunoabsorption test was done. Briefly, before applying Ab-9204 to electroblotted polyvinylidene difluoride membrane, Ab-9204 was mixed with 40 μM APP-C100 at 37°C for 1 h and centrifuged at 10,000 g for 1 h. Then the supernatant was applied to the membrane.
The animals were carefully handled in a humane fashion, and our experimental protocols met the U.S. Public Health Service standards (Guide for the Care and Use of Laboratory Animals, NIH, 1985).
RESULTS
Complete ischemia of the forebrain for 10 min produced consistent neuronal degeneration in the hippocampal region in all animals surviving at 7 days after the ischemic insult but not in the animals killed before 24 h after the insult. All CA1 pyramidal neurons were affected bilaterally, and neurons in CA2/3 and granule cells in the dentate gyrus were morphologically intact.
In the sham-treated hippocampus, perikarya and dendrites of CA pyramidal neurons and dentate granule cells were lightly stained with Ab-9204 (Figs. 2A and 3A). Four hours after the ischemia, the immunoreactivity of the neurons in all CA sectors and dentate gyrus was significantly enhanced (Fig. 2B), especially in the CA1 pyramidal neurons (Fig. 3B). APP-C100 shows granule-like accumulation in the perikarya of CA1 neurons 1 day after the ischemia (Fig. 3C), while the other neurons showed a homogeneous distribution (Fig. 2C). This granule-like accumulation was observed in CA1 neurons until 2 days after the ischemia but not in the other neurons. In the medial part of the CA1 sector, pyramidal neurons were dead and lost immunoreactivity 3 days after the ischemia. Seven days after the ischemia, all CA1 pyramidal neurons were dead and lost immunoreactivity (Fig. 3D), while CA3 neurons and dentate granule cells still kept moderate immunoreactivity (Fig. 2D). While glial cells did not show any immunoreactivity throughout the experiment, those in the CA1 sector showed moderate immunoreactivity 7 days after the ischemia (Fig. 3D). These glial cells proved to be astrocytes by GFAP staining in serial section (Fig. 3E). In the section stained by double immunolabeling, astrocytes showed both APP-C100 and GFAP immunoreactivities (data not shown). Each group showed consistent tendency of immunohistochemical pattern among animals except for two: One animal killed 4 h after ischemia showed little immunoreactivity as did sham-treated animals. One animal killed 1 day after ischemia showed only enhanced immunoreactivity in whole hippocampus but not granule-like accumulation.
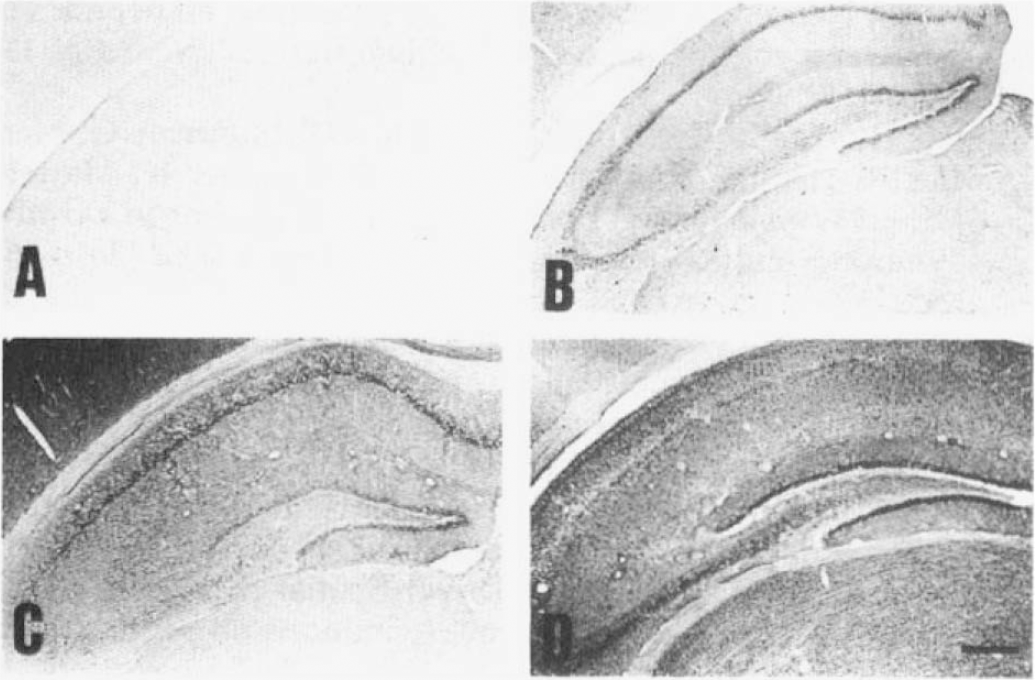
Immunohistochemical distribution of β-amyloid precursor protein (APP)-C100 in the hippocampus. In sham animals, APP-C100 was lightly observed in neurons of all CA sectors and dentate granule cells
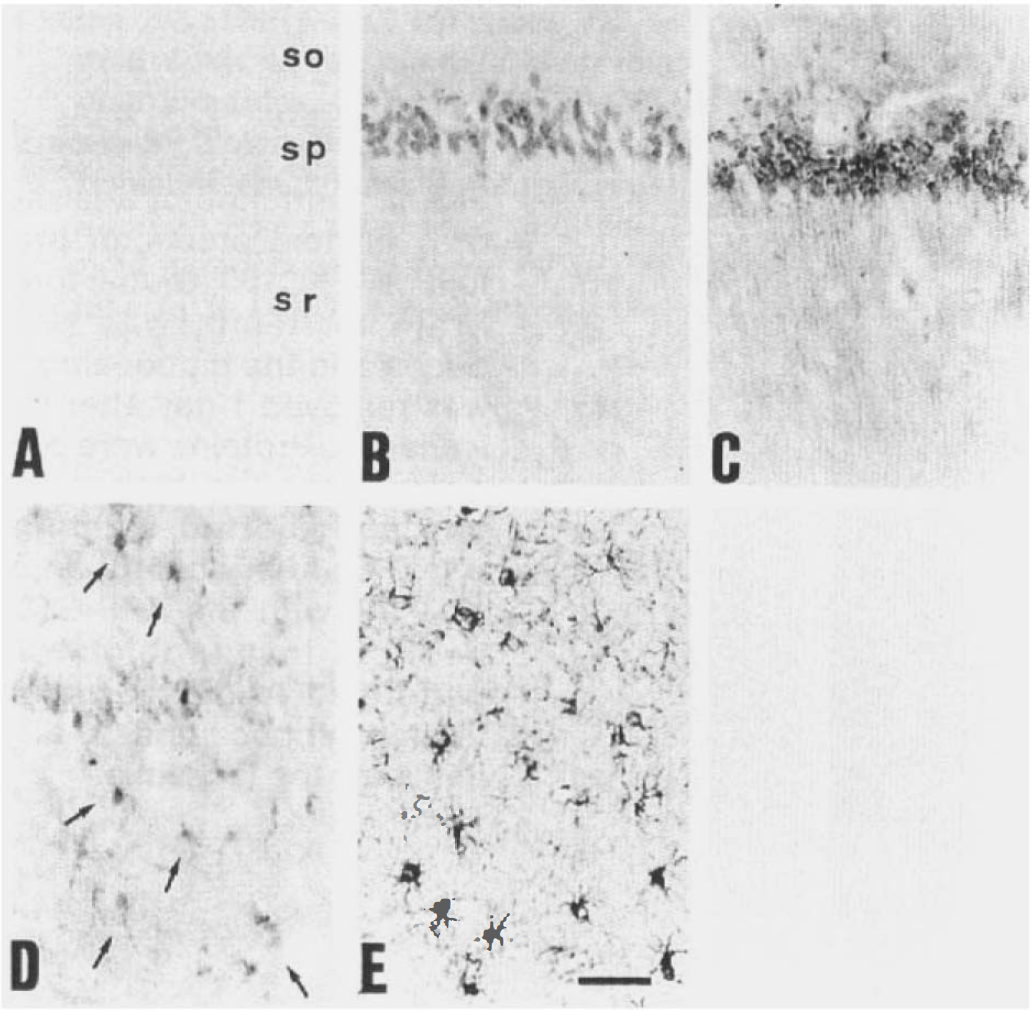
High power photomicrograph of CA1 sector. In sham animals, CA1 neurons showed light and homogeneous β-amyloid precursor protein (APP)-C100 immunoreactivity as well as neurons of the other CA sectors and dentate granule cells
The immunoblot study demonstrated that the immunoreactive substance in our immunohistochemical study using Ab-9204 was not Aβ (4 kDa) but APP-C100 (16 kDa), and 1 day after the ischemia, its expression was significantly enhanced (Fig. 4A) (% of sham density = 317 ± 35%; mean ± SD). The immunoreactivity on the Western blotting was completely eliminated by absorbing the Ab-9204 by excessive APP-C100 (Fig. 4B).
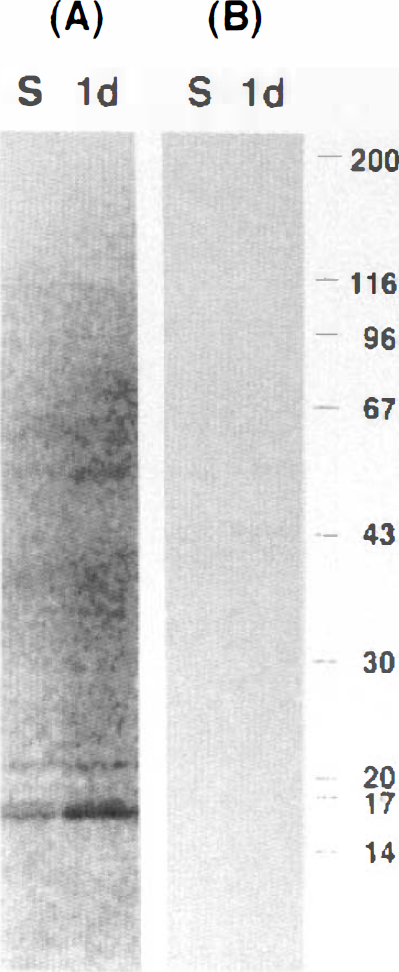
Immunoblot analysis of β-amyloid precursor protein (APP)-C100. Global forebrain ischemia was produced and the hippocampus was removed 1 day after the ischemia. Proteins were prepared from the hippocampus and blotted with Ab-9204
DISCUSSION
Deposition of Aβ in the form of neuritic plaques is one of the major pathological features of Alzheimer's disease (Mattson et al., 1993a; Mullan and Crawford, 1993; Selkoe, 1993), although Aβ-containing neuritic plaques are observed in other pathological conditions such as Down syndrome (Rumble et al., 1989). The transmembrane glycoprotein βAPP is enzymatically processed by α-secretase and liberates the physiologically secreted form from the cells. Alternative processing of βAPP by β-secretase can liberate Aβ comprising 4(M2 amino acids (Saido et al., 1995). APP-C100, which spans Aβ and cytoplasmic domains of the C-terminal fragment, is generated in embryonal carcinoma cells with full-length APP cDNA (Yoshikawa, 1993). The physiologically secreted form of βAPP has protective effect against ischemic or excitotoxic insults (Mattson et al., 1993b). On the other hand, aggregated forms of Aβ and possibly APP-C100 have a neurotoxic effect (Maruyama et al., 1990; Pike et al., 1993; Seubert et al., 1993). A recent study revealed that cultured brain cells secrete βAPP cleaved at the amino terminus of the Aβ (Seubert et al., 1993). Here we demonstrate for the first time that βAPP cleaved at the amino terminus of the Aβ (APP-C100) accumulates in vivo and is enhanced under the pathological condition of cerebral ischemia. It may be difficult to clarify the source of the APP-C100. However, Maruyama et al. (1990) demonstrated that APP-C100 is anchored on plasma membrane in COS cells overexpressing APP-C100 and it cannot be released to the extracellular space before cell death. In our study, significant accumulation of APP-C100 was observed in CA1 neurons 1 day after the ischemia when the neurons are still alive. Taken together, APP-C100 in CA1 neurons seems to be produced in these neurons. Because a small amount of APP-C100 was observed in the neurons of sham-treated animals, it might have a physiological function as does Aβ in the cultured neurons (Yanker et al., 1990). After the ischemic insult, accumulation of APP-C100 was enhanced in the hippocampal neurons. This may be secondary to neuronal degeneration. Ten minutes of forebrain ischemia induces delayed neuronal death in CA1 neurons, but the neurons are morphologically intact 24 h after the insult. Therefore, granule-like aggregation of APP-C100 possibly takes place as a primary event. Tomimoto et al. (1994) demonstrated that the secreted form of βAPP, which has a neuroprotective effect, accumulates in CA3 neurons after global forebrain ischemia but not in CA1 neurons. In our study, neurotoxic APP-C100 accumulated in neurons of all CA sectors and dentate granule cells. Accumulation of these two substances with opposite effects may produce selective vulnerability of hippocampal neurons after ischemia. Pike et al. (1991) demonstrated that the aggregated but not the monomeric form of Aβ showed neurotoxicity in rat hippocampal cultures. In the present study, granule-like aggregation of APP-C100 was observed only in CA1 neurons affected by ischemic insult. In the other neuronal cells, APP-C100 showed homogeneous distribution in the perikarya and dendrites throughout the experiment. While the relationship between APP-C100 and neurotoxicity still needs further documentation, this difference of APP-C100 conformations may play a role in development of delayed neuronal death. In astrocytes of the CA1 sector, accumulation of APP-C100 was observed 7 days after ischemia. In the chronic stage, ˜21 days later, glial cells proliferated in the CA1 sector. Therefore, accumulation of APP-C100 in astrocytes does not result in cell death of the astrocyte. APP-C100 may have a different biological effect in glia than in neurons.
Our results demonstrate that an ischemic insult modifies amyloidogenic APP processing in hippocampal neurons that display delayed cell death. This animal model may provide a clue to understanding the etiology of Alzheimer's disease and may have a potential role in the development of amyloid-inhibitory therapeutics.