
Abstract
We examined the effects of GABA receptor stimulation on the neuronal death induced by exogenously added excitatory amino acids or combined oxygen–glucose deprivation in mouse cortical cell cultures. Death induced by exposure to NMDA, AMPA, or kainate was attenuated by addition of GABA or the GABAA receptor agonist, muscimol, but not by the GABAB receptor agonist, baclofen. The antiexcitotoxic effect of GABAA receptor agonists was blocked by bicuculline or Picrotoxin. In contrast, GABA or muscimol, but not baclofen, markedly increased the neuronal death induced by oxygen–glucose deprivation. Muscimol potentiation of neuronal death was associated with increased glutamate efflux to the bathing medium, and increased cellular 45Ca2+ accumulation; it was blocked by MK-801, but not NBQX, suggesting mediation by NMDA receptors. Bicuculline only weakly attenuated muscimol potentiation of oxygen–glucose deprivation-induced neuronal death, probably because it itself increased this death. Present results raise a note of caution in the proposed use of GABAA receptor stimulation to limit ischemic brain damage in vivo.
Excessive extracellular accumulation of the excitatory neurotransmitter, glutamate, and subsequent excitotoxic neuronal death have been implicated in the pathogenesis of hypoxia–ischemic neuronal death (Benveniste et al., 1984; Simon et al., 1984; Meldrum, 1985; Choi and Rothman, 1990; Albers et al., 1992). More recently, attention has also focused on the possibility that the inhibitory neurotransmitter, γ-aminobutyric acid (GABA), may serve as a brake on overexcitation and thus reduce the pathogenesis of ischemic brain damage. Extracellular GABA levels are also increased during hypoxia-ischemia in vivo (Hagberg et al., 1985; Globus et al., 1988). The idea that this increase may counteract the toxic effects of glutamate led Globus et al. (1991) to propose that an “excitotoxicity index,” calculated as [glutamate] × [glycine]/[GABA] in the extracellular space, might better predict ischemic brain damage than glutamate levels alone. GABA inhibition is mediated both by GABAA receptors, which open membrane chloride channels and stabilize the membrane potential below firing threshold, and GABAB receptors, which act via G proteins to reduce transmitter release from presynaptic terminals (Kaila, 1994).
Recent studies from several laboratories have examined enhancement of GABA receptor activation as a strategy for reducing brain damage in animal models of ischemia. Several such strategies reduce hippocampal CA1 neuronal loss following global ischemia in gerbils: (a) preischemic administration of muscimol, baclofen, or diazepam (Sternau et al. 1989); (b) pre- or postischemic administration of chlormethiazole (Cross et al., 1991); (c) pre- and postischemic intraventricular infusion of a GABA-transaminase inhibitor or muscimol (Shuaib et al., 1992, 1993; see also Abel and McCandless, 1992, which utilized preischemic administration); and (d) postischemic administration of diazepam (Schwartz et al., 1994). In rats, pre- and postischemic administration of GABA uptake inhibitor or diazepam (Johansen and Diemer, 1991) or postischemic administration of diazepam (Schwartz et al., 1995) reduced hippocampal CA1 cell loss following global ischemia. Neurological function following multifocal ischemia in rats was improved by postischemic administration of muscimol (Lyden and Hedges, 1992; Lyden and Lonzo, 1994), and chlormethiazole pretreatment reduced infarct volume in a rat model of transient focal ischemia (Sydserff et al., 1995). Protective effects of barbiturates have also been observed in both focal and global ischemia models (see review by Spetzler and Hadley, 1989), although the multiplicity of known barbiturate actions hinders interpretation of these observations strictly in context of GABAA receptors.
In contrast to the consistent neuroprotective effects of GABAmimetic drugs in animal models of ischemia, both protective and injurious effects have been observed in studies of excitotoxicity in vitro. In our murine cortical cell cultures, neither GABA nor diazepam reduced the rapidly triggered excitotoxic neuronal death induced by a brief intense glutamate exposure (Koh and Choi, 1987b). Using a more prolonged and weaker N-methyl-
While this last observation could simply reflect distortions induced by cell culture methods, it is also possible that cell culture isolates an important deleterious effect of GABAA receptor activation that is masked in vivo by more powerful beneficial effects. Thus, we set out to characterize in detail the effects of both GABAA and GABAB receptor agonists on the neuronal death induced by excitotoxins or oxygen–glucose deprivation in cortical cell cultures. Abstracts have appeared elsewhere (Monyer et al., 1990; Muir et al., 1994; Muir and Choi, 1995).
MATERIALS AND METHODS
Cell culture
Primary cortical cell cultures containing both neuronal and glial elements were prepared from fetal mice at 15–16 days of gestation as previously described (Rose et al., 1993). Dissociated cortical cells were plated at approximately 2.75 hemispheres per 24-multiwell (15-mm) vessels (Primaria; Falcon) on a previously established glial bed (see below) in modified Eagle's minimal essential medium (MEM, Earle's salts, supplied glutamine-free) supplemented with 5% heat-inactivated horse serum, 5% fetal calf serum, glutamine (2 mM), and glucose (total 25 mM). Cultures were maintained at 37°C in a humidified incubator containing 5% CO2. Nonneuronal cell division was halted after 5–7 days in vitro by a 1- to 3-day exposure to 10 μM cytosine arabinosine. Cultures were subsequently fed twice weekly with a medium identical to the plating medium except lacking fetal serum, and used for experiments between 13 and 16 days in vitro. Glial cultures were prepared similarly from 1- to 3-day-old mice neocortices plated at 0.5–1 hemisphere per 24-multiwell vessel in a plating medium similar to that described above, but containing 10% heat-inactivated horse serum, 10% fetal calf serum, and 10 ng/ml epidermal growth factor.
Excitatory amino acid exposure
Rapidly triggered excitotoxicity induced by a 5-min exposure to 100 μM NMDA was carried out at room temperature in a HEPES-buffered salt solution containing (millimolar): 120 NaCl, 5.4 KCl, 0.8 MgSO4, 1.8 CaCl2, 20 HEPES, 15 glucose, and 0.01 glycine (pH 7.4). GABA or related drugs were applied concurrently with NMDA exposure. Cultures were then returned to MEM containing 10 μM glycine. Slowly triggered excitotoxicity was induced by a 24-h exposure to NMDA (15 μM), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA, 10 μM), or kainate (350 μM) in MEM supplemented with 10 μM glycine at 37°C. MK-801 (10 μM) was included with AMPA or kainate to block secondary activation of NMDA receptors.
Oxygen-glucose deprivation
Cultures were placed in an anaerobic chamber (Forma Scientific, Marietta, OH, U.S.A.) containing a gas mixture of 5% CO2, 10% H2 85% N2 (<0.2% O2) (Goldberg and Choi, 1993). Culture medium was replaced by thorough exchange with a deoxygenated, glucose-free balanced salt solution (BSS) containing (millimolar): 116 NaCl, 5.4 KCl, 0.8 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 1.8 CaCl2, and 0.01 glycine. Cultures were then placed in a 37°C humidified incubator within the chamber. Following 40–45 min oxygen–glucose deprivation, the exposure medium was exchanged with oxygenated MEM containing glucose and cultures returned to a normoxic incubator. Specified drugs were added during the period of oxygen–glucose deprivation only.
Cyanide exposure
Cultures were exposed to 1 mM potassium cyanide in oxygenated, glucose-free BSS at 37°C. Exposure was terminated by washout with oxygenated MEM containing glucose and cells returned to an incubator for 20–24 h. Specified drugs were added during the period of cyanide exposure only.
Assessment of neuronal injury
Neuronal cell death was estimated by examination of cultures under phase-contrast microscopy and quantitated by measurement of lactate dehydrogenase (LDH) released by damaged or destroyed cells in the bathing media 20–24 h after insult initiation (Koh and Choi, 1987a). LDH-mediated conversion of pyruvate to lactate was monitored by measuring levels of reduced nicotinamide adenine dinucleotide (NADH), by absorbance at 340 nm, using a spectrophotometric microplate reader (UVmax, Molecular Devices, Menlo Park, CA, U.S.A.). LDH was converted to units per milliliter by fitting the rate of change of absorbance of each sample to a LDH standard curve (Sigma Enzyme Control 2-E). The LDH signal corresponding to near-complete neuronal death without glial death was measured in sister cultures exposed to 300 μM NMDA for 24 h (total neuronal LDH). Since absolute total LDH levels may vary among plates obtained from different dissections, LDH values were normalized to the levels associated with control injury in sister cultures. Basal LDH levels determined in sister cultures subjected to sham wash (generally <15% of total LDH) were subtracted from values obtained in experiments to yield the signal specific to experimental injury.
45Ca2+ accumulation
Cultures were exposed to NMDA for 5 min or deprived of oxygen and glucose as described above with the inclusion of specified drugs and 45Ca2+ (New England Nuclear, Wilmington, DE, U.S.A.; final activity, 2 μCi/ml) in the exposure medium (Hartley et al., 1993; Goldberg and Choi, 1993). Exposure was terminated by thoroughly washing cells of extracellular 45Ca2+. Subsequently, the extracellular medium was removed, cells were lysed with 0.2% sodium dodecyl sulfate, and an aliquot was added to scintillation fluid for counting. Since absolute neuronal density may vary among plates obtained from different dissections, experimental values were normalized to the levels associated with control injury (= 100) in sister cultures. Previous study has determined that 45Ca2+ accumulation is mostly neuronal (Hartley et al., 1993).
High-performance liquid chromatography (HPLC)
Samples of the bathing medium collected prior to the termination of oxygen–glucose deprivation were assayed for glutamate by using phenylisothiocyanate (PITC) derivatization, HPLC separation using a Hypersil-ODS reverse-phase column (Hewlett Packard, Wilmington, DE, U.S.A.), and ultraviolet detection at a wavelength of 254 nm (Cohen et al., 1986). A 200 μl of sample medium was derivatized with 100 μl of PITC, methanol, and triethylamine (2:7:4) and dried under vacuum. The samples were then reconstituted in solvent consisting of 0.14 μM sodium acetate, 0.05% triethylamine, 6% acetonitrile, and brought to pH 6.4 with glacial acetic acid, prior to the HPLC run. The above solvent was used as the mobile phase with the column being washed with 60% acetonitrile/40% water between each sample run. Media glutamate concentrations were calculated by normalizing to glutamate standards. Glutamate measurements were found to be linear over the range 0.1–10 μM, with 0.1 μM being the threshold for reliable detection.
Reagents
Culture medium was purchased from Gibco (Grand Island, NY, U.S.A.) as a 10× concentrated stock lacking bicarbonate and glutamine; serum was obtained from Hyclone Laboratories Inc. (Logan, UT, U.S.A.). NMDA, GABA, muscimol hydro-bromide, Picrotoxin, and phenylisothiocyanate were obtained from Sigma Chemical (St. Louis, MO, U.S.A.). (R)-(+)-Baclofen hydrochloride, guvacine hydrochloride, bicuculline methiodide, nipecotic acid, and MK-801 were purchased from Research Biochemicals Inc (Natick, MA, U.S.A.).
RESULTS
Rapidly triggered excitotoxicity
Consistent with an earlier study (Choi et al., 1988), exposure of mixed neuron/glial cortical cell cultures to 100 μM NMDA for 5 min resulted in degeneration of a majority (50–75%) of the neuronal population 20–24 h later. Concurrent application of either 100–1000 μM GABA (Fig. 1A), or 3–100 μM muscimol (Fig. 1B), a specific GABAA receptor agonist, produced a partial, concentration-dependent reduction in NMDA-induced neuronal death. In contrast, the specific GABAB receptor agonist, baclofen, had no effect on NMDA-induced neuronal death over the tested concentration range of 1–100 μM (Fig. 1C). Application of 1 mM GABA, muscimol, or baclofen alone for 24 h was not toxic.
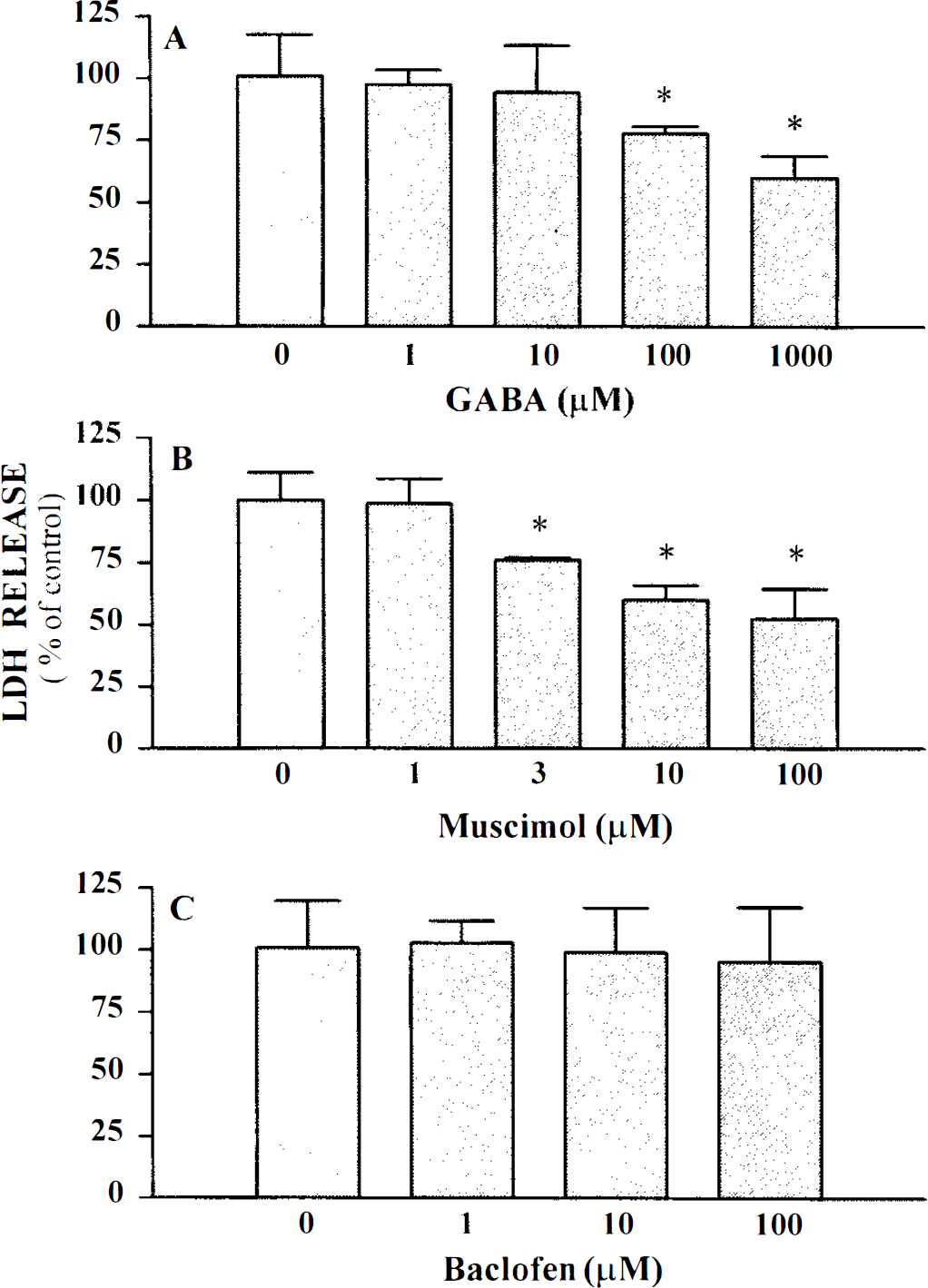
GABAA receptor agonists attenuate NMDA-mediated excitotoxicity. Cultures were exposed to 100 μM NMDA for 5 min in the absence or presence of increasing doses of GABA agonists. LDH was measured 20–24 h later. Data represent mean LDH ± SD scaled to the mean LDH measured in cultures exposed to 100 μl NMDA alone (=100).
NMDA-stimulated uptake of 45Ca2+, which correlates with subsequent neuronal death (Hartley et al., 1993), was partially reduced by 10–100 μM muscimol (Table 1). 45Ca2+ accumulation was completely blocked by the NMDA antagonist, MK-801 (10 μM).
Effect of muscimol on NMDA-stimulated 45Ca accumulation a

Cultures were exposed to 200 μM NMDA for 5 min in the presence of extracellular 45Ca2+ and the indicated concentration of muscimol or MK-801. Following exposure, cells were washed and lysed to measure intracellular (neuronal) 45Ca2+ accumulation. Data represent mean ± SD, n = 16–20 cultures per condition from five experiments, scaled to the mean values measured in cultures exposed to 200 μM NMDA alone (= 100).
Values are significantly different from NMDA alone by ANOVA followed by Dunnett's t test (p < 0.05).
To demonstrate that muscimol's protective action was mediated by GABAA receptor activation, specific antagonists for the receptor were utilized. The competitive GABAA receptor antagonist, bicuculline (10–100 μM), antagonized the protective effect of muscimol on NMDA-induced neuronal death (Fig. 2A). A similar antagonism was demonstrated with the noncompetitive GABAA receptor antagonist, picrotoxin (10–100 μM, Fig. 2B). Neither antagonist increased NMDA-induced neuronal death when applied alone (data not shown).
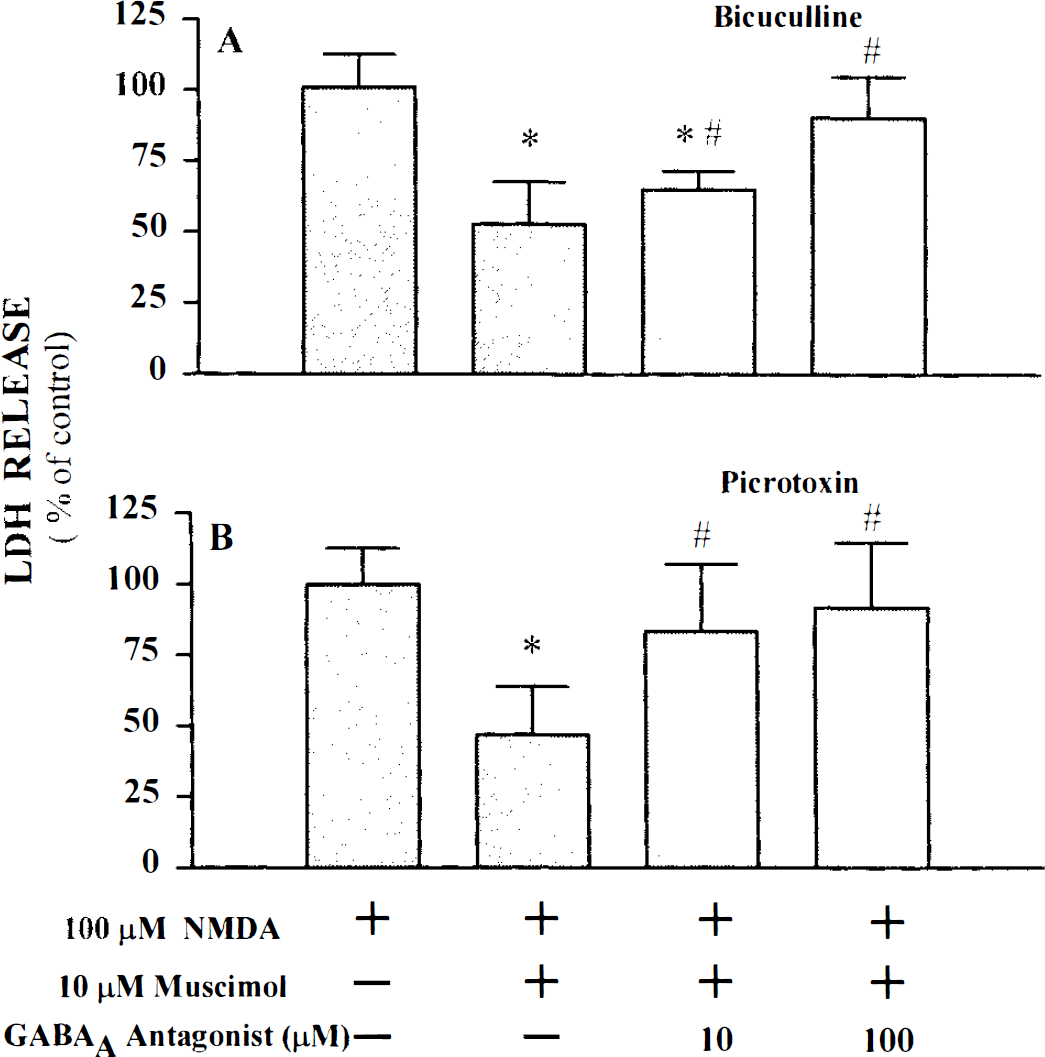
GABAA receptor antagonists block muscimol's protective action on NMDA excitotoxicity. Cultures were exposed to 100 μM NMDA for 5 min in the absence or presence of the indicated drugs. LDH was measured 20–24 h later. Data represent mean LDH ± SD scaled to the mean LDH measured in cultures exposed to 100 μM NMDA alone (=100).
Slowly triggered excitotoxicity
In addition to reducing rapidly triggered excitotoxicity, muscimol also reduced the slowly triggered excitotoxic euronal loss induced by a 24-h exposure to 15 μM NMDA, 10 μM AMPA, or 35–40 μM kainate (Table 2). A concentration-dependent reduction in neuronal death was observed with each agonist, although the protective effect against AMPA- or kainate-induced death was weaker than that against NMDA-induced death (Table 2, Fig. 1B). Reduction of NMDA-induced death was also observed with 1 mM guvacine or nipecotic acid, inhibitors of GABA uptake, but not against AMPA or kainate injury (Table 2). Addition of baclofen did not affect NMDA-, AMPA-, or kainate-induced neuronal death (data not shown).
Effect of muscimol or GABA uptake inhibitors on slowly triggered excitotoxicity by NMDA, kainate, or AMPA a
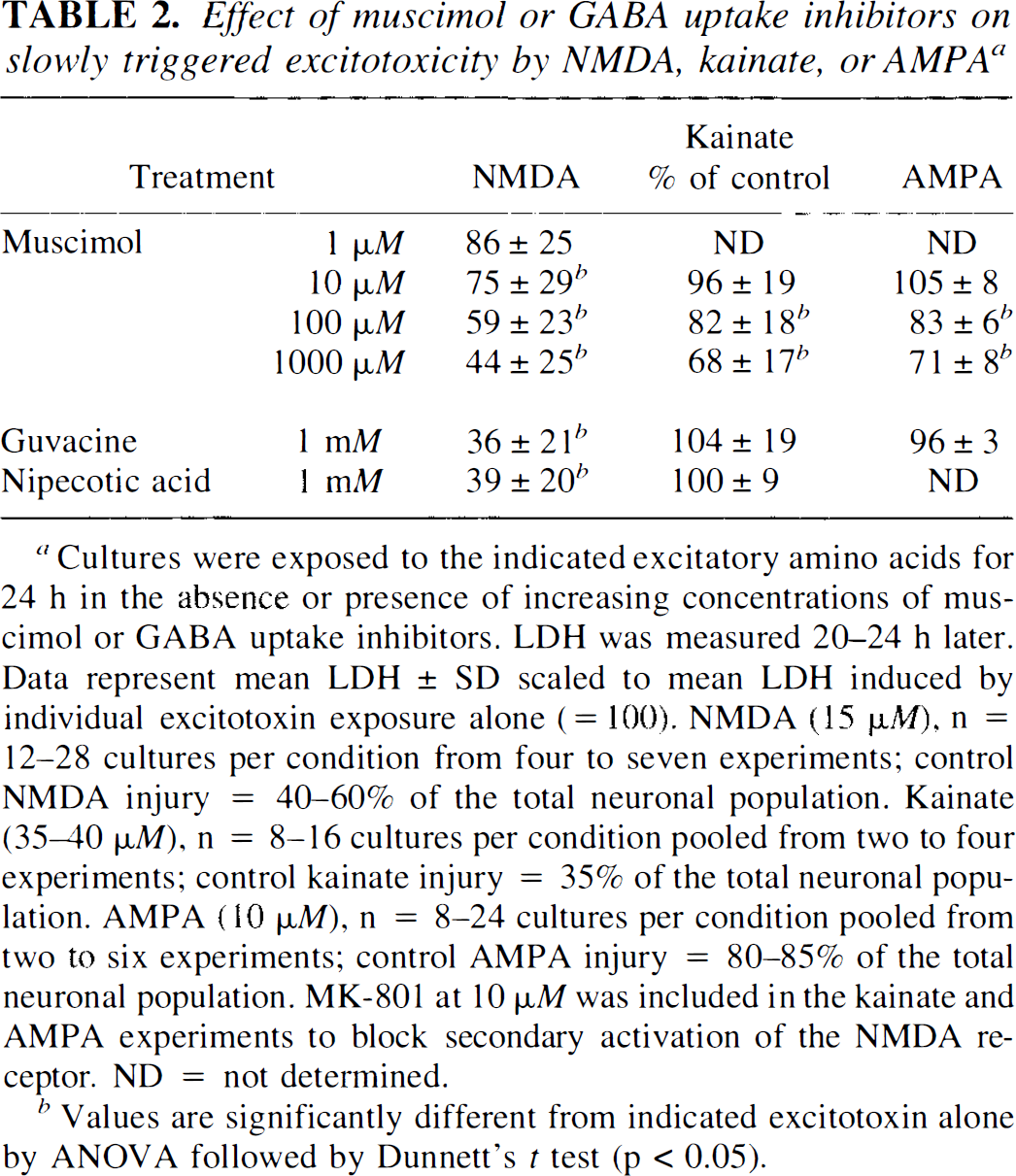
Cultures were exposed to the indicated excitatory amino acids for 24 h in the absence or presence of increasing concentrations of muscimol or GABA uptake inhibitors. LDH was measured 20–24 h later. Data represent mean LDH ± SD scaled to mean LDH induced by individual excitotoxin exposure alone (= 100). NMDA (15 μM), n = 12–28 cultures per condition from four to seven experiments; control NMDA injury = 40–60% of the total neuronal population. Kainate (350 μM), n = 8–16 cultures per condition pooled from two to four experiments; control kainate injury = 35% of the total neuronal population. AMPA (10 μM), n = 8–24 cultures per condition pooled from two to six experiments; control AMPA injury = 80–85% of the total neuronal population. MK-801 at 10 μM was included in the kainate and AMPA experiments to block secondary activation of the NMDA receptor. ND = not determined.
Values are significantly different from indicated excitotoxin alone by ANOVA followed by Dunnett's t test (p < 0.05).
Oxygen-glucose deprivation
Consistent with earlier studies (Goldberg and Choi, 1993; Koh et al., 1995), a 40- to 45-min period of oxygen and glucose deprivation produced a low level (10–30%) of neuronal death 20–24 h later. In striking contrast to the protective effect observed for excitotoxicity, addition of 10–100 μM GABA or 1–100 μM muscimol (but not baclofen) during this oxygen–glucose deprivation period produced a concentration-dependent increase in immediate neuronal swelling and an exacerbation of neuronal death 20–24 h later (Fig. 3). With 100 μM GABA or muscimol, the same 40- to 45-min period of oxygen–glucose deprivation induced the death of a majority of neurons. This GABA- or muscimol-induced potentiation of neuronal death after oxygen–glucose deprivation was blocked by 10 μM MK-801 (Fig. 3). An antagonist of AMPA/kainate receptors, NBQX (30 μM) had no effect.
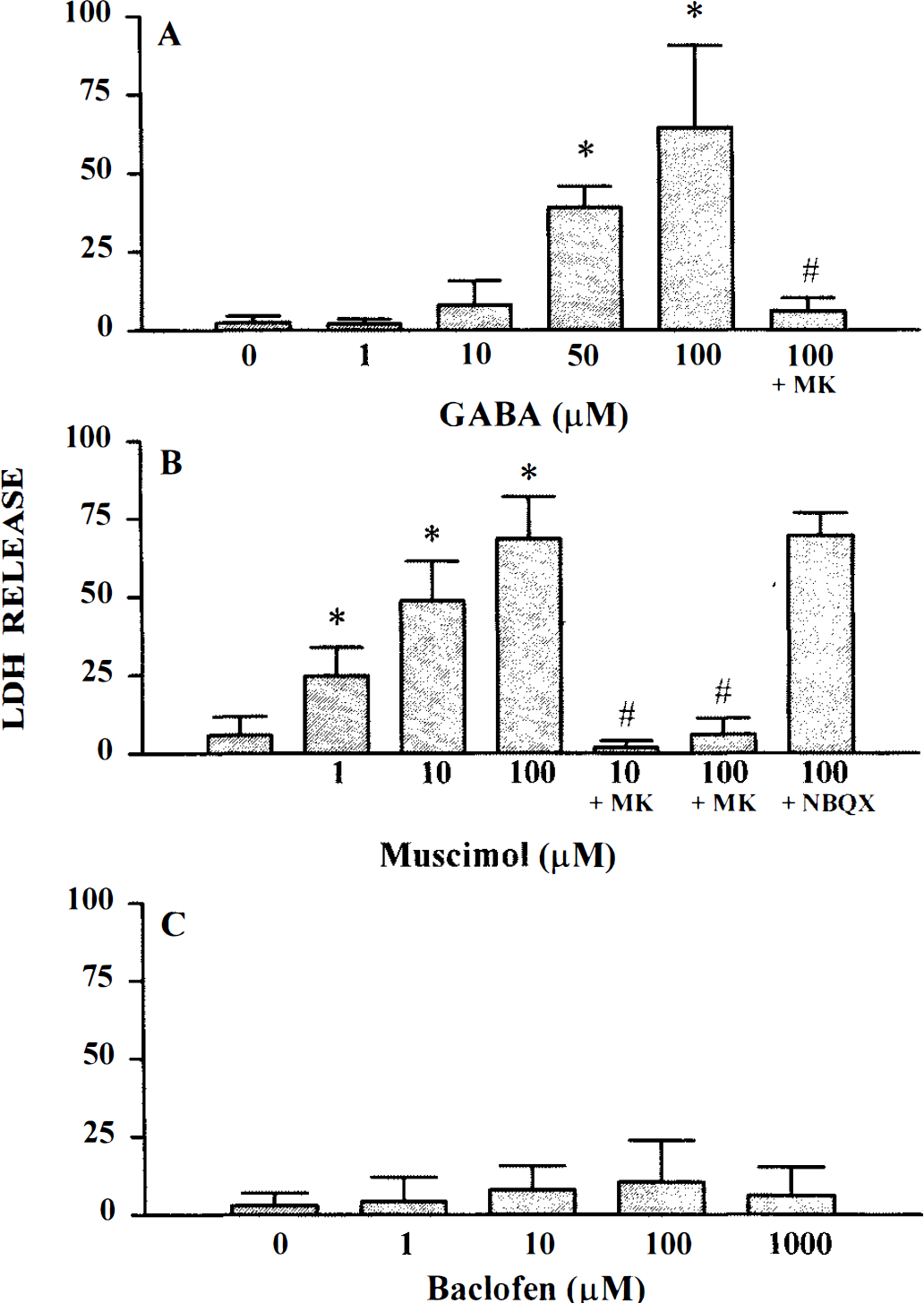
GABAA receptor agonists potentiate neuronal injury induced by oxygen–glucose deprivation. Cultures were deprived of oxygen and glucose for 40–45 min in the absence or presence of the indicated drugs. LDH was measured 20–24 h later. Data represent mean LDH ± SD expressed as percentage of total neuronal LDH.
Addition of 1–100 μM muscimol also markedly increased accumulation of glutamate in the extracellular medium, and intracellular 45Ca2+ uptake, measured immediately after a 40- to 45-min period of oxygen–glucose deprivation (Fig. 4). Muscimol itself did not alter intracellular 45Ca2+ uptake in sham-washed control cells. MK-801 at 10 μM attenuated 45Ca2+ accumulation but did not significantly decrease accumulation of extracellular glutamate (Fig. 4). Bicuculline (1–100 μM) only weakly reduced the muscimol-induced potentiation of neuronal death after oxygen–glucose deprivation (Fig. 5A), probably because it alone produced a concentration-dependent, MK-801-sensitive, increase in this neuronal death (Fig. 5B). Similar results were obtained using Picrotoxin (data not shown).
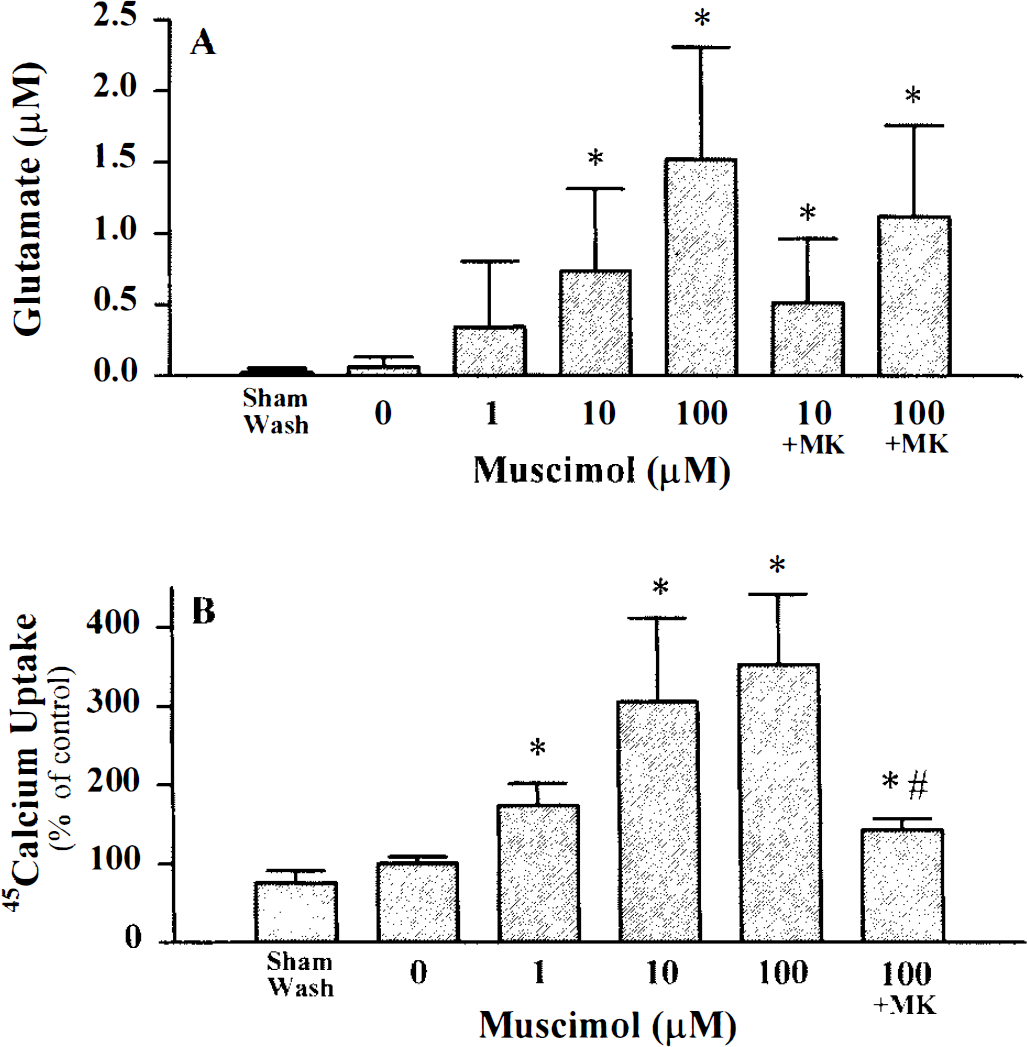
Effect of muscimol on glutamate release and 45Ca2+ accumulation induced by oxygen–glucose deprivation. Cultures were deprived of oxygen and glucose for 40–45 min in the presence of the indicated concentration of muscimol.
Cyanide exposure
As an additional neuronal injury paradigm triggered by energy depletion, cultures were exposed to cyanide. GABA or muscimol addition also potentiated the neuronal death induced by energy-depletion due to a 40- to 45-min exposure to 1 mM potassium cyanide in glucose-free media (Fig. 6).
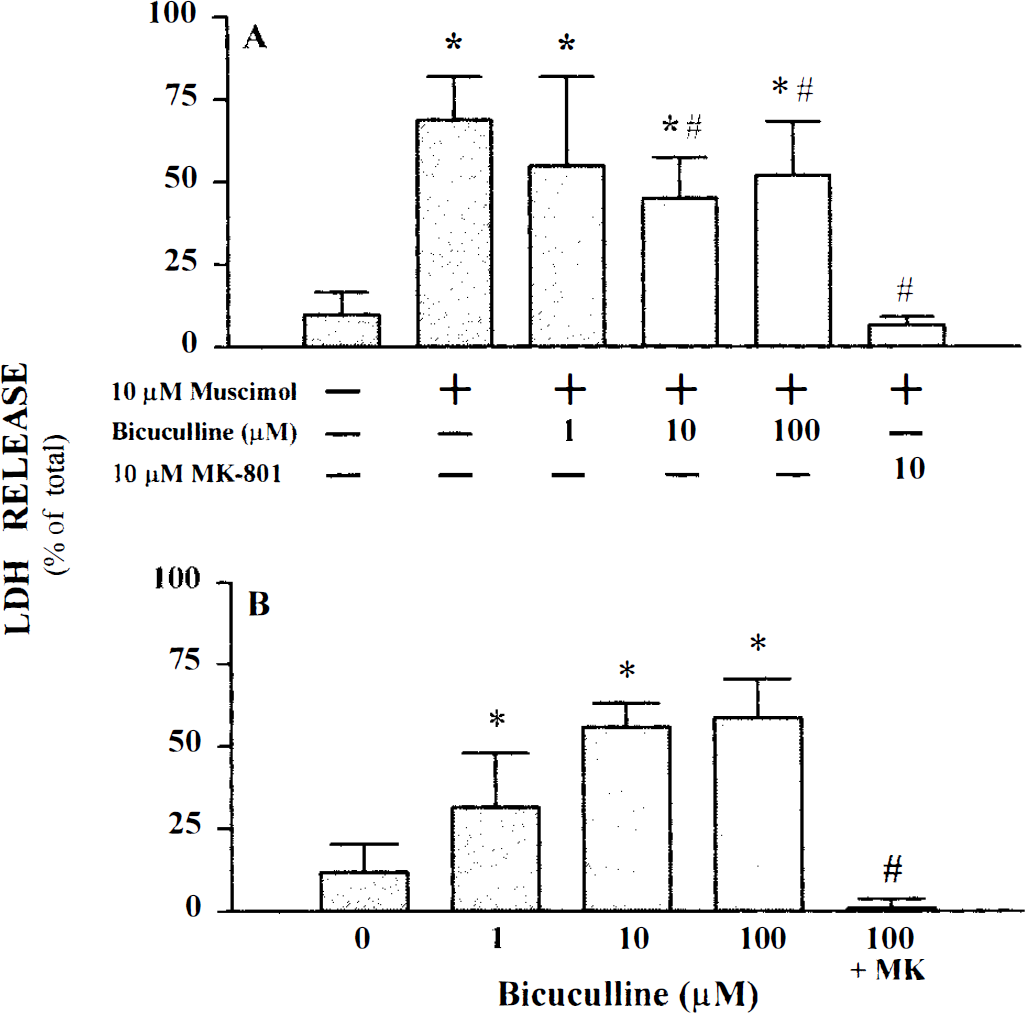
Effect of bicuculline on neuronal injury induced by oxygen–glucose deprivation in the presence and absence of muscimol. Cultures were deprived of oxygen and glucose for 40–45 min in the absence or presence of the indicated drugs. LDH was measured 20–24 h later. Data represent mean LDH ± SD expressed as percentage of total LDH.
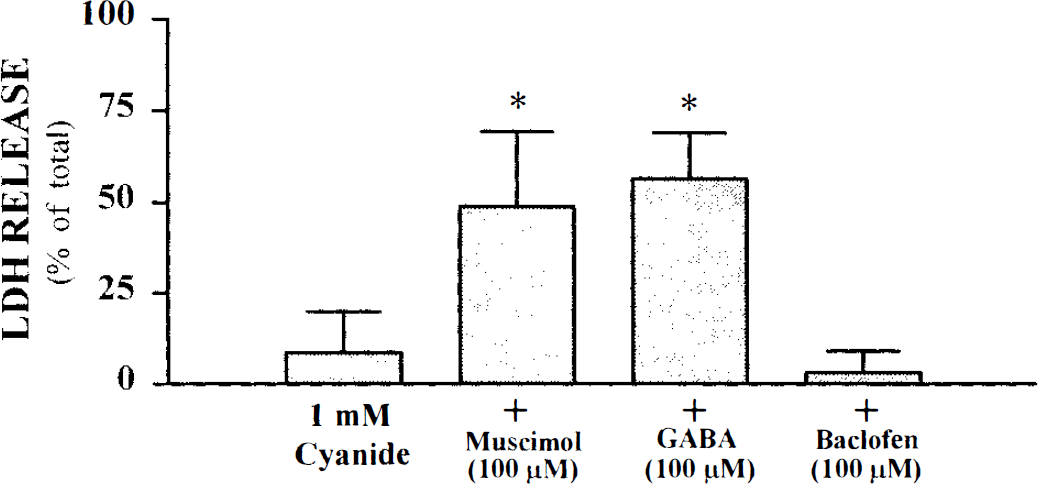
GABAA receptor agonists potentiate neuronal injury induced by cyanide exposure. Cultures were exposed to potassium cyanide for 40–45 min in the absence or presence of the indicated drugs. LDH was measured 20–24 h later. Data represent mean LDH ± SD expressed as percentage of total LDH; n = 16 cultures per condition pooled from four experiments. An asterisk indicates values significantly greater from cyanide exposure alone as determined by ANOVA followed by Dunnett's t test (p < 0.05).
DISCUSSION
Present data document an unexpected contrast between the protective effect of GABAA agonists on the cortical neuronal death induced by exogenous NMDA addition and the exacerbating effect of the same drugs on the neuronal death induced by either oxygen–glucose deprivation or cyanide exposure. The latter is unexpected for two reasons: (a) there is convincing evidence that GABAmimetic drugs are neuroprotective in several animal models of global or focal ischemia (see above); and (b) prior study in our system has indicated that the death induced by short periods of oxygen–glucose deprivation (such as used here) is mostly mediated by NMDA receptors (Goldberg and Choi, 1993).
In earlier studies, neither we (Koh and Choi, 1987b; Monyer et al, 1990) nor Erdo and Michler (1990) observed a neuroprotective effect of GABA agonists against maximal levels of rapidly triggered, NMDA receptor-mediated excitotoxicity. Detection of a partial protective effect of GABA agonists in the present study probably reflects employment of lower NMDA concentrations, which produced submaximal neuronal death. Our observations are consistent with the finding by Ohkuma et al. (1994) that muscimol reduced the neuronal injury of cultured cortical neurons induced by low (10 μM) concentrations of NMDA. Taken together, available data suggest that GABAA receptor stimulation produces a partial protective effect against submaximal NMDA receptor-mediated toxicity that can be overridden by intense NMDA receptor activation.
The protective effect of muscimol and GABA versus excitatory amino acid toxicity may be due to the ability of GABAA receptor activation to drive the membrane potential towards Ec1, which is usually near the resting potential. This should increase the voltage-dependent Mg2+ block of the NMDA receptor-gated channel (Mayer et al., 1984; Nowak et al., 1984) and thereby reduce NMDA receptor-mediated toxic Ca2+ entry (Fig. 7). In addition, membrane hyperpolarization can be expected to reduce Ca2+ influx mediated via the Na+-Ca2+ exchanger or mediated via voltage-gated Ca2+ channels (Choi, 1988). Consistent with the idea that GABAA receptor activation reduces NMDA-induced net neuronal Ca2+ influx, it decreased cellular accumulation of extracellular 45Ca2+ induced by a 5-min exposure to 200 μM NMDA. Reduction of depolarization-induced Ca2+ influx may also explain the ability of GABAA receptor activation to attenuate the slowly triggered excitotoxic neuronal death induced by a 24-h exposure to NMDA, AMPA or kainate.
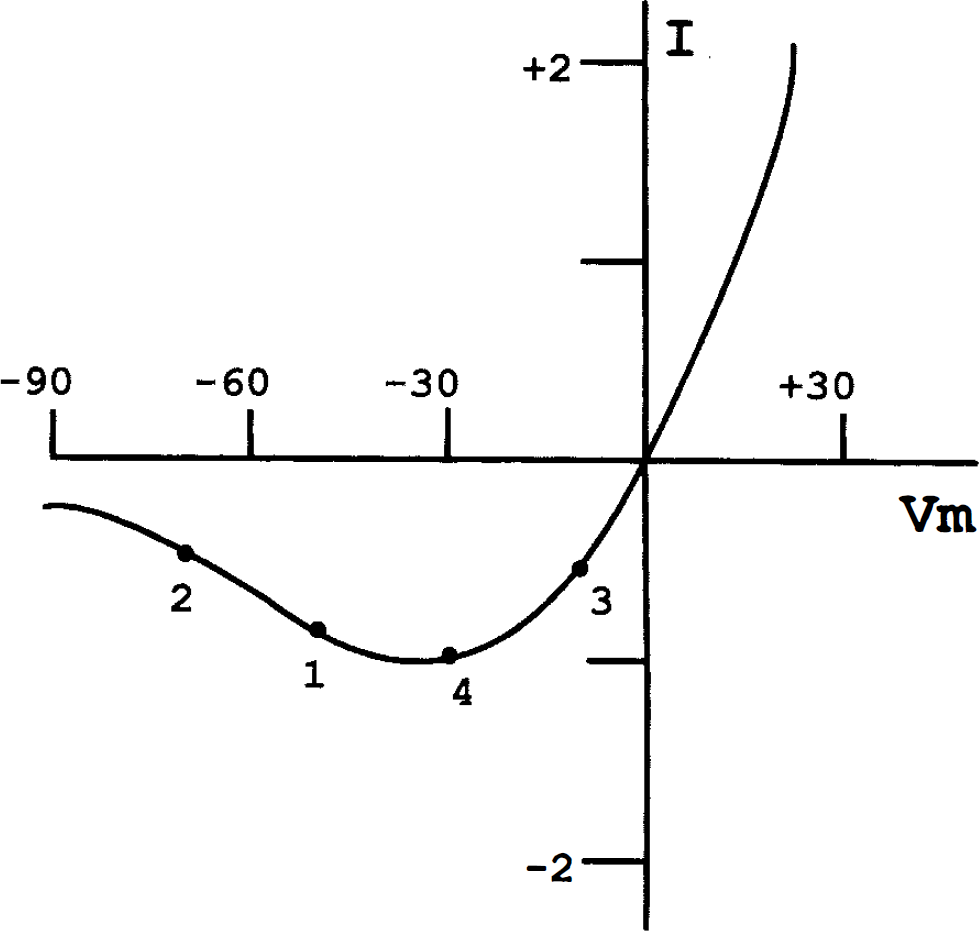
Proposed effect of GABAA receptor activation on NMDA receptor-mediated current (I) in energy-competent and energy-depleted neurons. This graph depicts a typical NMDA receptor I/V curve, exhibiting voltage-dependent Mg2+ block (modified from MacDermott and Dale, 1987). In energy-competent neurons (only partially depolarized by exposure to NMDA), the hyperpolarizing effect of GABAA receptor activation may move the membrane potential from point 1 to point 2, thereby strengthening the Mg2+ block. However, in energy-depleted neurons, the same GABAA receptor activation may move membrane potential from point 3 to point 4, producing no effect on the Mg2+ block, but still increasing the inward driving force for Ca2+.
In slowly triggered excitotoxicity paradigms, addition of either muscimol or drugs that inhibit GABA uptake produced a greater protective effect on NMDA-induced neuronal death than on AMPA- or kainate-induced death. Perhaps this is because the effect of membrane hyperpolarization on the Mg2+ block of NMDA receptor-gated channels is not shared with AMPA or kainate receptor-gated channels. The observed lack of protective effect of GABA uptake inhibitors against AMPA or kainate toxicity likely reflects lower levels of GABAA receptor stimulation achieved via this route (compared to that achieved during NMDA exposure), perhaps because of less GABA release.
Further study will be required to elucidate the mechanisms underlying the unexpected potentiating effect of GABAA receptor activation on oxygen–glucose deprivation-induced neuronal death. Data presented here indicate that this potentiated death is associated with enhanced Ca2+ accumulation and is still mediated by NMDA receptors. One intriguing possible explanation can be developed from the idea that membrane hyperpolarization has opposing effects on NMDA receptor-mediated Ca2+ influx: increasing the Mg2+ block of the NMDA receptor-gated channel, but also increasing the electrochemical gradient favoring Ca2+ entry. Thus, in energy-competent neurons with hyperpolarized membrane potentials (e.g., neurons exposed to exogenously added NMDA), GABAA receptor activation may move the membrane potential towards Ec1 enough that the Mg2+ block is enhanced and Ca2+ influx through the NMDA receptor-gated channel falls (Fig. 7). However, in energy-depleted neurons with collapsed membrane potentials (e.g., neurons exposed to oxygen–glucose deprivation or cyanide; see Hansen, 1985), GABAA receptor activation may hyperpolarize neuronal membranes enough to increase the inward Ca2+ driving force, but not enough to bring in the Mg2+ block (Fig. 7). Increased neuronal Ca2+ entry may also lead to greater glutamate release and help explain the observed increase in extracellular glutamate accumulation observed in cultures exposed to oxygen–glucose deprivation in the presence of muscimol. Increased extracellular glutamate itself would be expected to increase neuronal death.
Other mechanisms can also be considered. For instance, although GABAA receptor stimulation usually inhibits central neuronal firing, it can, under some circumstances, be excitatory (Michelson and Wong, 1991; Kaila, 1994). In particular, while normally inhibitory, dendritic GABAA receptors can become excitatory during intense activation due to an activity-dependent shift in the GABAA reversal potential away from Ec1 and toward EHCO3 (Staley et al, 1995). Furthermore, it is conceivable that prolonged exogenous application of GABA or muscimol desensitizes GABAA receptors sufficiently that a paradoxical loss of inhibitory tone results. The apparent voltage-dependence of this desensitization (Frosch et al., 1992) raises the possibility that desensitization might be greater when the agonists are applied prior to membrane depolarization (as would be the case in the oxygen–glucose deprivation and cyanide paradigms), compared with when the agonists are applied together with a depolarizing stimulus (as would be the case in the excitotoxicity paradigms).
The weak antagonism of bicuculline or picrotoxin on the injury-potentiating effect of muscimol in cultures deprived of oxygen and glucose is well explained by the observed injury-potentiating effect exhibited by these GABAA antagonist drugs when applied alone. This intrinsic injury-potentiating effect may reflect an effect of GABAA receptor antagonists to increase circuit excitability, enhancing ATP depletion and glutamate release. In exogenous excitotoxicity induced by addition of NMDA, endogenous glutamate release is likely swamped by exogenously added NMDA.
In contrast to the observed modulatory effects of GABAA receptor activation upon cortical neuronal death induced by exposure to excitotoxins or oxygen–gluocse deprivation, GABAB receptor activation with baclofen lacked effect. The picture appears different in CNS white matter, which has few glutamate receptors and is not vulnerable to excitotoxic injury. Fern et al. (1995) recently reported that anoxia-induced injury of the rat optic nerve was attenuated by GABA or baclofen, but not GABAA receptor agonists.
The present in vitro experiments should not be viewed as conflicting with existing in vivo data indicating that GABAA agonists can reduce ischemic brain injury. The final weighting of different variables can only be accomplished in vivo, and nothing demonstrated here argues against a final predominance of beneficial effects. However, present results provide novel isolation of a potential deleterious effect of GABAA receptor activation upon energy-depleted neurons. Strategies aimed at eliminating this deleterious action—for example, addition of NMDA receptor blockade (see for example, Lyden and Lonzo, 1994)—might further enhance the neuroprotective effects of GABAA agonists in the ischemic brain.
Footnotes
Abbreviations used
Acknowledgment:
We thank Susan Adams and Aghdas Jafari for technical support and Dr. Sandra Hewett for helpful discussions. This work was supported by NIH research grant NS-32636 (D.W.C.).