
Abstract
A series of experiments was performed to determine the role of interleukin (IL)-1 in the induction of tolerance to global ischemia in Mongolian gerbils. In Group I, a 2-min “preconditioning” ischemia protected CA1 hippocampal neurons in gerbils subjected to 3.5 min ischemia 3 days later. CA1 neuronal density was: sham, 171 ± 3/mm; 3.5 min ischemia, 30 ± 30/mm; 2 and 3.5 min ischemia 162 ± 6/mm. Experiments in Group II addressed the role of IL-1 in the induction of tolerance by sublethal ischemia. Arterial IL-1α and IL-Iβ became elevated between 1 and 3 days after a 2-min ischemic exposure. IL-1α was: sham, 6.4 ± 0.6 ng/ml; and 2-day, 10.2 ± 1.2 ng/ml. IL-1β was: sham, 6.4 ± 0.5 ng/ml; and 2-day, 17.3 ± 2 ng/ml. Recombinant human IL-1 receptor antagonist (IL-1ra) i.p. blocked ischemic tolerance induction by 2-min preconditioning ischemia: 2-min ischemia + vehicle, 162 ± 6/mm; and 2-min ischemia + IL-1ra, 67 ± 17/mm. Experiments in Group III assessed the capacity of IL-1 to induce tolerance to brain ischemia. IL-1α i.p. (0, 10, 20 μg/kg) for 3 days prior to 3.5-min forebrain ischemia provided significant CA1 neuroprotection in a dose-dependent manner: 2 ± 2, 68 ± 83, and 129 ± 42/mm, respectively. IL-1β (15 μg/kg) in combination with either IL-1ra (100 mg/kg) or IL-1ra vehicle i.p. on the same schedule demonstrated a significant CA1 neuroprotection that could be nullified by IL-1ra: IL-1β + IL-1ra vehicle, 153 ± 16/mm; and IL-1β + IL-1ra, 67 ± 36/mm. Recognition that tolerance arises from stimulation of a known receptor (IL-1RI) permits molecular analysis of the intracellular signaling that is critical for production of that state.
Current efforts to develop effective stroke therapy are generally directed at arresting progressive brain damage in the ischemic penumbra (Ginsberg and Pulsinelli, 1994; Hossmann, 1994). Although a variety of experimental therapies aimed at counteracting one mediator or another demonstrate significant neuroprotection in animal models, they tend to perform weakly, if at all, in clinical trials of acute stroke in humans. One possible explanation for the difficulties encountered in efforts to develop an effective stroke treatment is that the conventional search for a single or dominant mediator of progressive ischemic brain damage is incompatible with the essential nature of the problem. The multiplicity of factors that participate in progressive ischemic brain injury and their intricate interconnections, in the aggregate, can produce dramatic tissue damage. Each factor by itself, however, may have only minor pathophysiologic importance (Hallenbeck and Dutka, 1990; Hallenbeck and Frerichs, 1993).
It is very difficult to deduce, by analyzing the interrelationships among the various causal factors, an effective intervention for a problem that stems from a constellation of minor causes. The need for such a deductive approach may be circumvented by studying states in which animals become tolerant to brain ischemia (Frerichs et al., 1994). Identification of the adjustments made by animals during natural or induced states of tolerance could guide investigators toward an understanding of effective therapeutic principles that would be difficult to discern through an analysis of mediator cascades. The goal would be to identify the intracellular signalling step(s) that can globally modulate the mediator network that causes progressive ischemic brain damage. In a sense, animals under these circumstances may be able to teach investigators how to solve this complicated problem.
Brief durations of “preconditioning” ischemia and hyperthermia have been observed to induce remarkable tolerance against subsequent, more severe ischemia that would otherwise lead to cell death in hippocampal CA1 neurons (Kitagawa et al., 1990; Kirino et al., 1991; Kitagawa et al., 1991b). However, no previous reports have clearly defined what is responsible for the initiation and development of such tolerance. If a ligand-receptor interaction could be identified that induced tolerance, it would greatly facilitate characterization of the intracellular signaling pathways that regulate the tolerant state. Interleukin (IL)-1 is a key immunomodulator that acts rapidly and mediates a wide range of immune and inflammatory responses as well as the acute phase reaction (Oppenheim et al., 1986). It is specifically antagonized by a recombinant human IL-1 receptor antagonist (IL-1ra), M.W. 18 kDa that has no apparent agonist function (Lennard, 1995). Stresses such as brain ischemia and surgical brain trauma have been shown to induce marked expression of IL-1 in neurons and microglia (Minami et al., 1992; Liu et al., 1993; Tchelingerian et al., 1993). Although neurons possess IL-1 receptors in the hippocampus (Ban et al., 1991), receptor-mediated effects of this cytokine in the brain are just beginning to be studied. Inhibition of IL-1 activity and receptor binding have been shown to attenuate ischemic and excitotoxic neuronal damage and edema formation (Relton and Rothwell, 1992; Martin et al., 1994; Garcia et al., 1995; Yamasaki et al., 1995). The present work is based on the hypothesis that exposure to IL-1 induces resistance to ischemia, which would otherwise lead to neuronal cell death, in the gerbil hippocampus.
METHODS
Mongolian gerbils (Meriones unguiculatus), 13–15 weeks of age, were subjected to the 18 experiments that make up this study. The experiments cluster into three groups. Experiments in Group I (Table 1) demonstrate the ability of a preconditioning exposure to sublethal ischemia to induce tolerance to a subsequent neuron-damaging ischemia (n = 20). Experiments in Group II (Table 2) address the role of IL-1 in the induction of tolerance by sublethal ischemia (n = 64). Experiments in Group III (Table 3) assess the capacity of IL-1 to induce tolerance to brain ischemia (n = 55). All experimental procedures in the present work were reviewed and approved by the National Institutes of Health Animal Care and Use Committee.
Group I. Ischemic tolerance
Animals were anesthetized with 1.5% halothane in 30% O2/70% N2O (vol/vol). Both common carotids were exposed through a midcervical incision and gently separated from their carotid sheaths. In sham-operated animals, both common carotids were exposed but not occluded. In experimental animals, both common carotids were simultaneously occluded with miniature vascular aneurysmal clips for either 2 or 3.5 min to produce transient global ischemia of the forebrain. Body temperature was monitored with rectal and temporal muscle thermometers and temporal muscle temperature was regulated at 37.2 ± 0.1°C by a feedback-controlled heating lamp and pad during surgery. After removal of the vascular clips, restoration of blood flow was verified by direct observation through a microscope. Rectal temperature was monitored following clip removal during the first 2 h of reperfusion. Surgical incisions were sutured and disinfected; animals were allowed free access to food and water, and were housed at constant temperature (23°C) under diurnal light conditions. To produce ischemic tolerance, gerbils were initially exposed to “preconditioning” ischemia for 2 min and were then subjected to a second, potentially neuron-damaging ischemia for 3.5 min 3 days later. These experiments are depicted in Table 1.
In order to determine whether 2 min of preconditioning ischemia increased circulating IL-1 in the blood, serum contents of IL-1α and IL-1β were measured by competitive enzyme immunoassay (Cytokit Red m1α and m1β, respectively) (Cyto-Immune Sciences, College Park, MD, U.S.A.). The anti-murine antibodies in these assays are known to cross react with rabbit and rat IL-1, and the sensitivity of the assay is 0.19 ng/ml. Blood (200 μl) was withdrawn from the common carotid artery (under pentobarbital anesthesia, 40 mg/kg, i.p.) from control and experimental animals at 3 h, 1,2, and 3 days after 2-min of ischemia or sham operation. Recombinant human IL-1 receptor antagonist (IL-Ira), kindly donated by Amgen (Boulder, CO, U.S.A.), was dissolved in 10 mM sodium citrate buffer, pH 6.8, with 150 mM NaCl, 0.5 mM EDTA, and 0.1% Tween 80. Following sham operation or 2 min of ischemia, gerbils received a daily i.p. injection of either IL-1ra (100 mg/kg) or the IL-Ira vehicle in a volume of 0.5 ml/kg at 0 (30 min after the procedure), 1 or 2 days before undergoing 3.5 min of ischemia on day 3. Surviving hippocampal CA1 neurons were counted on day 10. These experiments are depicted in Table 2.
Group II. IL-1 in ischemic tolerance

Vra, vehicle for IL-ra. The number of animals sampled at each time point was as follows: 3 h, 5; 1 day, 5; 2 days, 6; 3 days, 5.
Recombinant murine IL-1α, 2.4 × 106 U/μg (Sigma Chemical, St. Louis, MO, U.S.A.) was dissolved in 10 mM phosphate-buffered saline (PBS), pH 7.4, with 0.1% bovine serum albumin (endotoxin free). Recombinant human IL-1β, 1.9 × 104 U/μg containing 2.5 endotoxin units/mg protein (a gift from Dr. Craig W. Reynolds, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD, U.S.A.) was dissolved in 10 mM PBS, pH 7.4. The capacity of IL-1 to induce tolerance to ischemia was investigated by administering IL-1α (10 or 20 μg/kg) or its vehicle i.p. in a volume of 0.5 ml/kg on days 0, 1, and 2 before subjecting gerbils to 3.5 min of ischemia on day 3 and counting intact CA1 eurons on day 10. Another group of gerbils received daily i.p. IL-1β (15 μg/kg) or IL-1β vehicle in combination with either IL-1ra (100 mg/kg) or IL-1ra vehicle on each of 3 days prior to 3.5 min of ischemia on day 3. Intact CA1 neurons were counted on day 10. The experimental design is summarized in Table 3.
Group III. IL-1 induction of tolerance

V1α, V11β, and Vra denote vehicles for IL-1α, IL-1β, and IL-1ra, respectively; IL-1α(10), IL-1α 10 μg/kg and IL-1α(20), IL-1α 20 μg/kg.
Gerbils were killed by decapitation under deep pentobarbital anesthesia (100 mg/kg, i.p.) 7 days after ischemia. Their brains were immersion-fixed in ethanol/glacial acetic acid solution (19:1, vol/vol) at 4°C for 4 h, and embedded in paraffin. Five μm-thick coronal sections containing dorsal hippocampi were cut and stained in 0.1% cresyl violet solution. The number of intact neurons in the entire CA1 sector of the hippocampus was determined per mm of CA1 pyramidal cell layer and expressed as neuronal density, as described earlier (Kitagawa et al., 1990; Kirino et al., 1991; Kitagawa et al., 1991a).
All values reported here are expressed as mean ± SD. Overall statistical significance for differences among groups was tested by Kruskal-Wallis analysis. Comparisons between each group were made by the Dunnett-type nonparametric multiple comparison test. Values of p < 0.05 were considered-to be significant.
RESULTS
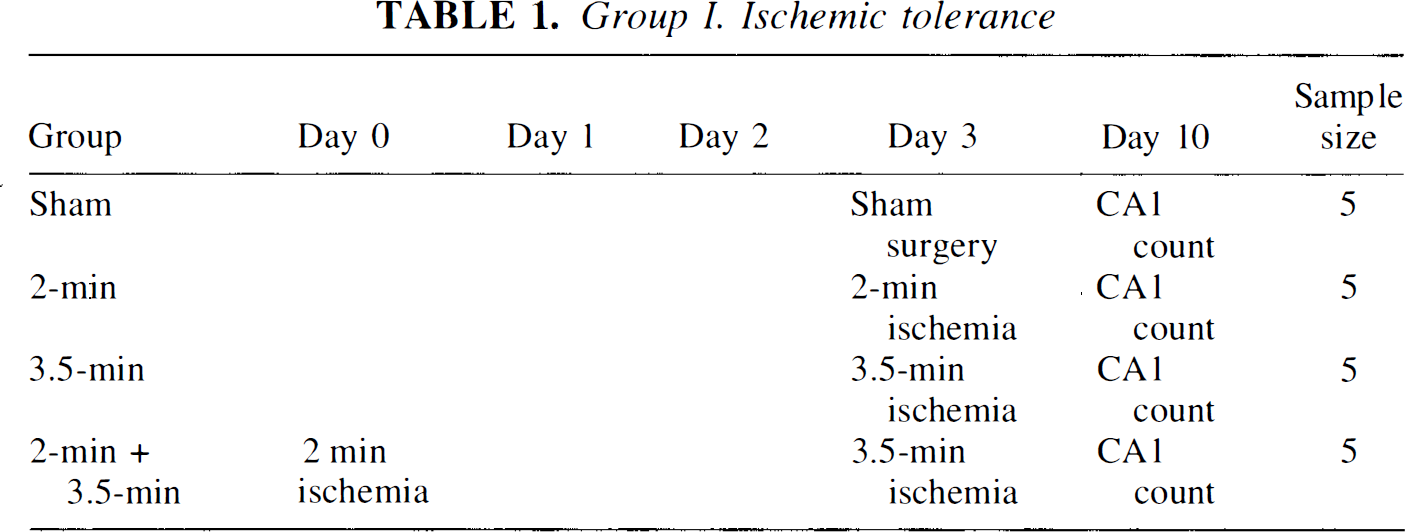
There was no significant difference in postischemic temperatures among the animals in the present study. Specifically, during the first several hours after injection of IL-1a, IL-1β, or vehicle, average body temperature rose only slightly, 0.1–0.4°C, with no difference between groups. After 3.5 min of ischemia, temperature rose 1.2–1.6°C, with no difference between the same groups. None of the gerbils showed epileptic motor activity. In sham-operated animals, no neuronal damage was evident in the CA1 region of the hippocampus 7 days after the procedure (CA1 neuronal density, 171 ± 3/mm). Transient ischemia for 2 min also caused no detectable damage (167 ± 3/mm), but 3.5-min of ischemia caused extensive neuronal destruction in the hippocampal CA1 sector measured 7 days later (30 ± 30/mm). In most animals subjected to preconditioning ischemia for 2 min followed by 3.5 min of ischemia 3 days later, CA1 neurons were spared (162 ± 6/mm) (Fig. 1).
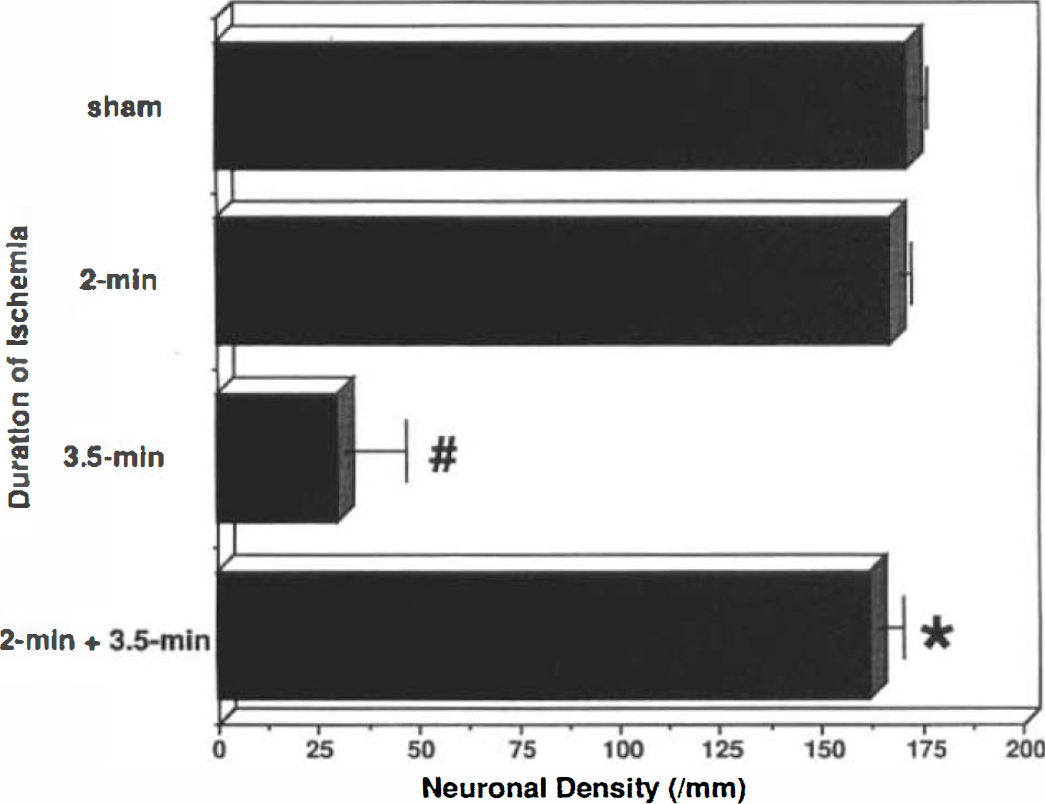
Effect of different durations of ischemia on delayed neuronal death in the hippocampal CA1 sector. #, p < 0.05 compared with sham-operated animals, *, p < 0.05 compared with animals subjected to a single 3.5-min period of ischemia (3.5-min). Error bars represent SDs in all figures.
The stress of surgery led to a small but significant elevation of circulating IL-1α and IL-1β that was sustained between 3 h and 3 days after the procedure, compared with controls. Serum contents of both IL-1α and IL-1β were significantly increased 1–3 days after exposure to 2 min of ischemia as compared to control animals and sham-operated animals (IL-1α: control, 4 ± 0.1 ng/ml; sham, 6.4 ± 0.6 ng/ml; and 2 days, 10.2 ± 1.2 ng/ml. IL-1β: control, 3 ± 0.7 ng/ml; sham, 6.4 ± 0.5 ng/ml; and 2 days, 17.3 ± 2 ng/ml) (Fig. 2).
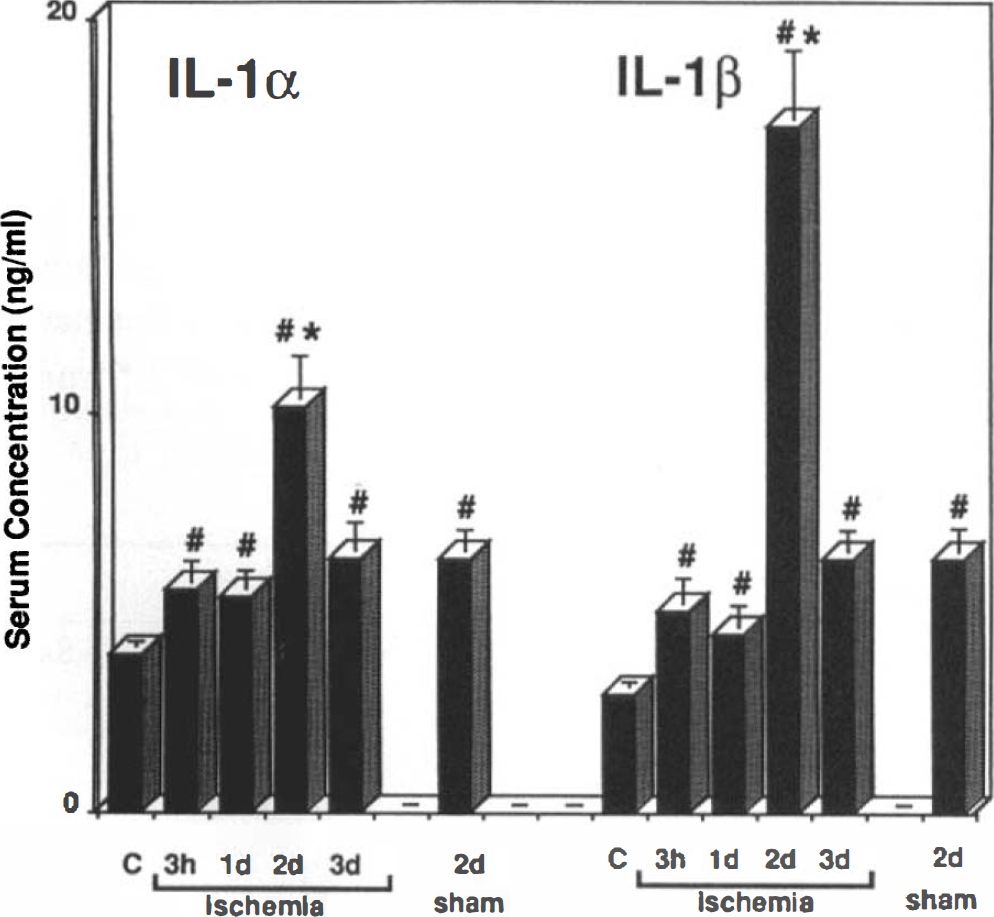
Serum concentration of IL-1α and IL-1β at several time points after 2 min of ischemia or after sham operation. #, p < 0.05 compared to nonischemic controls (C), *, p < 0.05 compared to sham-operated animals.
Gerbils were subjected either to a sham operation or a 2 min preconditioning ischemia followed by daily injections of vehicle or IL-1ra that started 30 min after the procedure. After 3 days, animals underwent 3.5 min of potentially neuron-damaging ischemia (Fig. 3). Sham operation followed by three daily injections of vehicle (sham + vehicle) offered no CA1 neuroprotection during the subsequent 3.5 min period of ischemia (22 ± 23/mm). A sham operation plus IL-1ra (100 mg/kg) injected 30 min later (day 0), and 1 and 2 days before 3.5 min of ischemia on day 3 (sham + IL-1ra) showed a trend toward neuroprotection that did not reach statistical significance (49 ± 32/mm). Preconditioning ischemia followed by daily vehicle injections provided clear-cut CA1 neuroprotection during a subsequent 3.5-min period of ischemia (2 min + vehicle: 162 ± 6/mm). IL-1ra, injected 30 min (day 0), and 1 and 2 days after a 2-min period of preconditioning ischemia, inhibited the expected neuronal protection in animals subjected to a subsequent 3.5 min of ischemia (2 min + IL-1ra: 67 ± 17/mm).
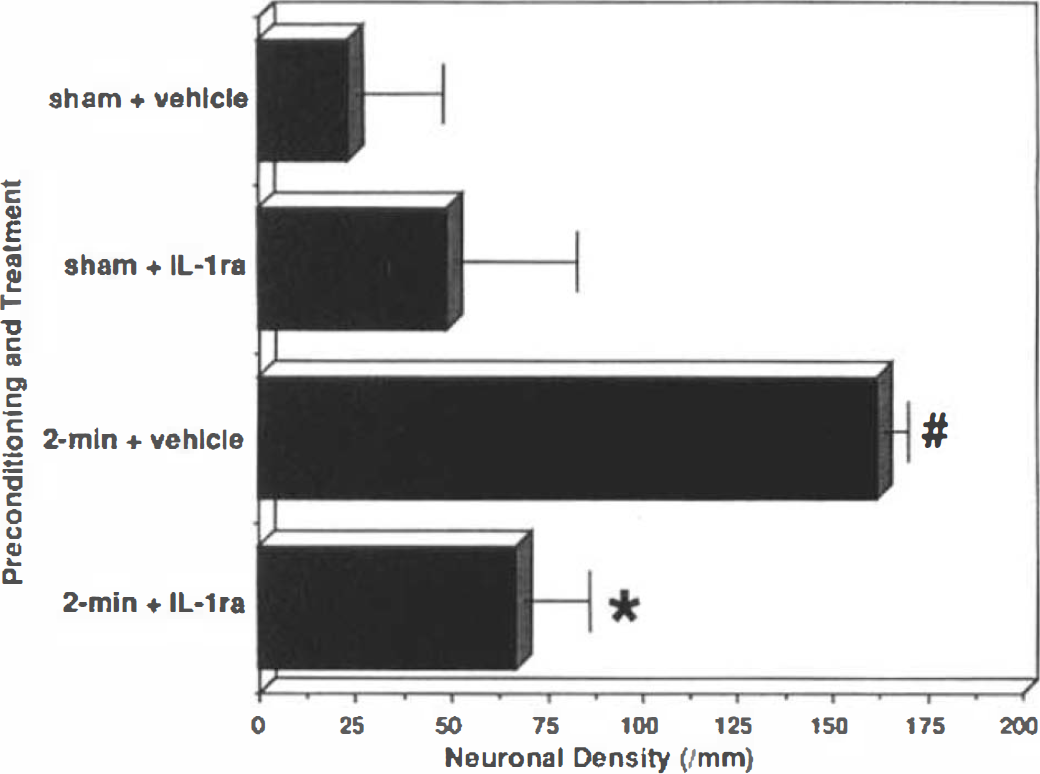
Effect of IL-1ra on CA1 neuronal tolerance to 3.5 min of forebrain ischemia induced by preconditioning ischemia for 2 min. #, p < 0.05 compared with sham-operated animals, *, p < 0.05 compared with “2-min + vehicle.”
Daily injection of IL-1α over a 3-day period protected CA1 hippocampal neurons from 3.5 min of global ischemia in a dose-dependent manner as compared with IL-1α vehicle injections. Neuronal counts were 2 ± 2/mm for vehicle, 68 ± 83/mm for IL-1α (10 μg/kg), and 129 ± 42/mm for IL-1α (20 μg/kg). No CA1 neuronal death was noted in five of the eight animals treated with IL-1α (20 μg/kg) (Fig. 4).
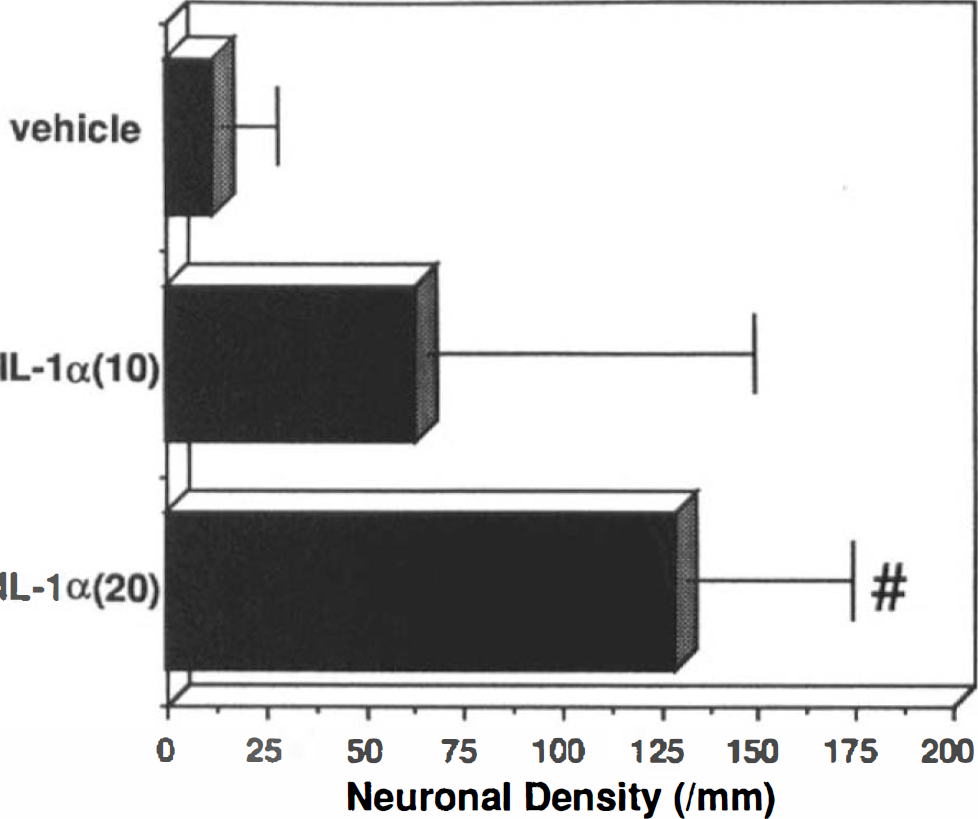
Effect of a daily schedule of IL-1α 0 (vehicle), 10 or 20 μg/kg i.p. × 3 on CA1 neuronal tolerance to a subsequent 3.5-min ischemia. #, p < 0.05 compared with animals administered vehicle.
IL-1β was also observed to induce ischemic tolerance, as demonstrated in Fig. 5. A combined daily injection of the vehicles for IL-1β and IL-1ra on each of 3 days prior to 3.5 min of ischemia did not reduce the loss of CA1 neurons after ischemic exposure (vehicle + vehicle: 23 ± 22/mm). IL-1β combined with the vehicle of IL-1ra on the same daily injection schedule provided marked protection with greater than 90% of CA1 neurons surviving 3.5 minutes of ischemia (IL-1β-vehicle: 153 ± 16/mm). A combination of IL-1β vehicle with IL-1ra (vehicle + IL-1ra) injected daily for 3 days prior to 3.5 min of ischemia was associated with a slightly higher average count of surviving CA1 neurons than the combined vehicle injection (49 ± 32/mm), but this difference did not reach statistical significance. The protection afforded by IL-1β plus IL-1ra vehicle was greatly reduced when IL-1β was co-injected with IL-1ra (IL-1β — IL-1ra: 67 ± 36/mm).
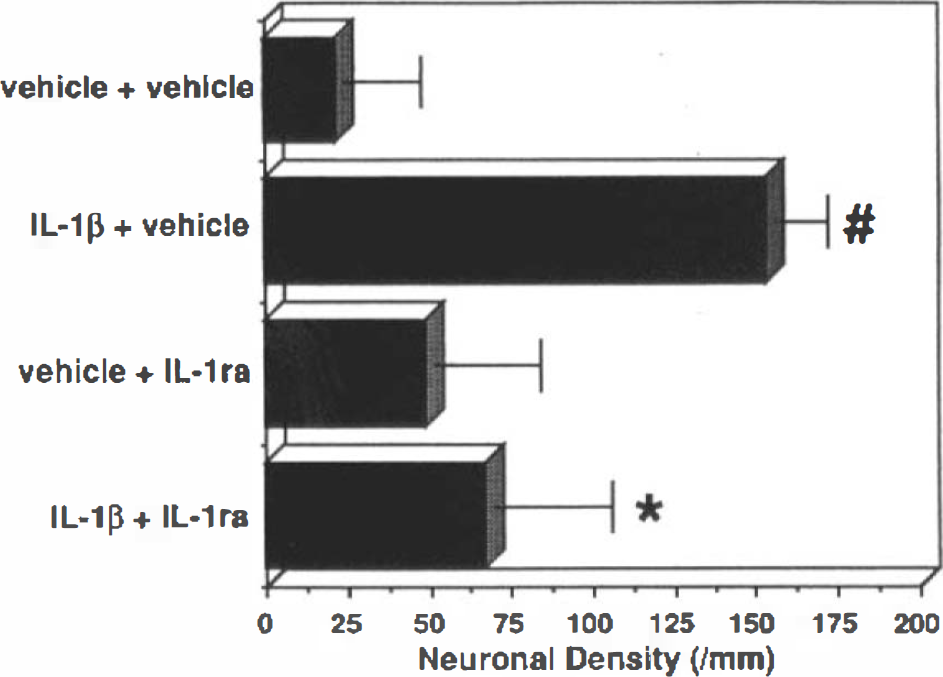
Effect of a daily schedule of IL-1β 0 (vehicle) or 15 μg/kg in combination with IL-1ra 0 (vehicle) or 100 mg/kg i.p. × 3 on CA1 neuronal tolerance to a subsequent 3.5 min period of ischemia. #, p < 0.05 compared with “vehicle + vehicle,” *, p < 0.05 compared with “IL-1 + vehicle.”
DISCUSSION
In this study, serum levels of both IL-1α and IL-1β increased between 1 and 3 days after sublethal ischemia produced by 2 min of bilateral common carotid artery occlusion in gerbils. This sublethal ischemia protected CA1 neurons from an otherwise lethal ischemia of 3.5 min 3 days later. Administration of IL-1ra blocked induction of ischemic tolerance in animals subjected to such sublethal ischemia. Both IL-1α and IL-1β were found to substitute for the sublethal ischemia with respect to induction of ischemic tolerance, and this effect was blocked by IL-1ra. Results demonstrate that IL-1 is necessary for induction of tolerance to neuron-damaging ischemia by prior exposure to sublethal ischemia (preconditioning) and that IL-1 is sufficient to directly and specifically induce tolerance to 3.5 min of global forebrain ischemia in the gerbil via a ligand-receptor interaction. Since the dosages of IL-1 used in this study did not provide total CA1 protection, we cannot distinguish (without an extensive dose-response design to optimize the IL-1 effect) between the possibility that IL-1 is the only stimulus required for the development of full ischemic tolerance and the possibility that IL-1 is a major factor, acting in concert with other factors. Incomplete suppression by IL-1 ra of ischemic tolerance produced by preconditioning ischemia or IL-1 is attributed to the capacity of IL-1ra to attenuate direct IL-1-mediated ischemic damage as an independent effect (Relton and Rothwell, 1992; Martin et al., 1994; Garcia et al., 1995; Yamasaki et al., 1995).
IL-1 administration 1–3 days prior to injury has been shown to protect hearts from ischemia/reperfusion injury and to reduce hyperoxic lung damage (White et al., 1987; Brown et al., 1990; Mauliket al., 1993). Several reported effects of IL-1 could contribute to its capacity to produce a state of ischemic tolerance in animals: IL-1 induces acute down-regulation of IL-1RI mRNA in perivascular cells (Ericsson et al., 1995); stimulates production of IL-1ra and tumor necrosis factor binding protein, which can inhibit the release and activity of IL-1 and TNF-α, respectively (Bargetzi et al., 1993; Marsh et al., 1994); and can also increase production of manganese superoxide dismutase, which could counteract damage in brain due to reactive oxygen species (Masuda et al., 1988). Other possible protective mechanisms involve release of antiinflammatory and anticoagulant cytokines such as transforming growth factor-β, IL-10, IL-4 (Jungi et al., 1994; Frankenberger et al., 1995) and inhibition of apoptosis by Bcl-2 effects on IL-1β-converting enzyme (ICE) (Chen et al., 1995). IL-1 is induced in brain by ischemia (Minami et al., 1992; Liu et al., 1993) and systemically by hyperthermia (Blake et al., 1994). Elaboration of this “alarm hormone” (Schöbitz et al., 1994) could be a generic mechanism for tolerance induction by stressful stimuli.
This work does not identify the cells directly affected by IL-1 binding to their receptors nor the consequent intracellular signaling that confers the ischemic brain tolerance. Circulating leukocytes, endothelium, and events at the blood-endothelial interface are strongly influenced by cytokines (reviewed in Hallenbeck, 1996). In addition, tolerance to lipopolysaccharide (LPS), a state mediated by cytokines, involves macrophages (Freudenberg and Galanos, 1988). These considerations raise the possibility that the observed tolerance arises from mechanisms that act systemically. Although penetration of the blood-brain barrier by IL-1 is, in general, low (Schöbitz et al., 1994), systemic IL-1 may recruit IL-1 expression in brain, as has been observed after systemic LPS administration (Gabellec et al., 1995) particularly in the region of the circumventricular organs (Breder et al., 1994). Systemic IL-1 may also signal brain cells indirectly by cytokine-receptor interaction in the vasculature and other blood-brain-barrier-related sites (Ericsson et al., 1995). Systemic administration of IL-1 can alter CBF and disrupt the blood-brain barrier (reviewed in Schöbitz et al., 1994). These considerations suggest that the observed tolerance could also arise from mechanisms that act within the brain.
The steps that culminate in the development of tolerance in this experimental paradigm proceed from an identifiable initiation point in contrast, for example, to experiments in which tolerance is induced by a bout of sublethal ischemia. The signaling receptor for IL-1 is the 80 kDa glycoprotein, IL-1RI. The other IL-1 receptor, a 68 kDa IL-1RII, has a very short intracytoplasmic domain comprised of 29 amino acids (in contrast to 215 amino acids of IL-1 RI) and does not appear to transmit intracellular signals so that it functions instead as a “decoy” or inhibitor of IL-1 (Brooks and Mizel, 1994; Saklatvala, 1995). The several modes of signal transduction from IL-1RI include the sphingomyelinase cycle, with its production of ceramide; protein kinases, e.g., p54 MAP kinase, β-casein kinase, hsp 27 kinase; phosphatases; and such transcription factors as API and NFκB (O'Neill, 1995). Identification of the critical steps for tolerance induction that proceed from this initiation point may permit a molecular basis for the regulation of tolerance to be determined.
Footnotes
Abbreviations used
Acknowledgment:
The authors wish to thank Mrs. Mary Crawford for secretarial assistance. Dr. Toshiho Ohtsuki is partially supported by the Japan Heart Foundation.