
Abstract
Stroke trials are initiated after demonstrated pharmacological protection in animal models. NBQX protects CA1 neurons against global ischemia; however, this glutamate antagonist induces a period of subnormal temperature (e.g., a decrease of only 1.0–1.5°C) lasting several days. In this study, NBQX (3 × 30 mg/kg, i.p.) was administered starting 60 min after reperfusion, and brain temperature had declined significantly below vehicle-treated animals by 2 h after reperfusion. When the postischemic brain temperature of NBQX-treated gerbils was regulated, no neuronal protection was found. Mimicking an NBQX-induced temperature profile for 28 h postischemia yielded histological protection 4 days later comparable to that of NBQX. However, both the NBQX and temperature simulation groups showed decreased protection after 10-day survival. Our data suggest that a protracted period of subnormal temperature during the postischemic period can obscure the interpretation of preclinical drug studies.
Delayed cell death resulting from cerebral ischemia is thought to be ultimately due to an excessive rise in free Ca2+ within vulnerable neurons, triggered by the excitatory neurotransmitter glutamate (Choi, 1988; Siesjö and Bengtsson, 1989). Initial optimism that N-methyl-D-aspartate (NMDA) receptor antagonists might be effective in treating stroke (Gill et al., 1987) has been tempered by the finding that the most potent of these compounds, MK-801, produced its neuroprotective action by inducing hypothermia (Buchan and Pulsinelli, 1990; Corbett et al., 1990; Nellgård et al., 1991). More recently, the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) antagonist, NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline), was reported to provide robust protection against neuronal loss in several animal models, even with postischemic treatment (Sheardown et al., 1990; Buchan et al., 1991; Diemer et al., 1992; Nellgård and Wieloch, 1992; Sheardown et al., 1993). Accordingly, it has been concluded that the AMPA receptor has a greater involvement in global ischemic injury than the NMDA receptor, and stroke trials with AMPA antagonists have been suggested (Buchan et al., 1993).
However, in the majority of studies in which NBQX has provided protection, temperature was not controlled during the postischemic period (Sheardown et al., 1990; Diemer et al., 1992; Sheardown et al., 1993). In view of the fact that many pharmacological studies of cerebral ischemia have been confounded by drug-induced hypothermia, it is essential that putative neuroprotective agents are examined closely to rule out similar interactions with brain temperature. In addition, recent data suggest that the preservation of CA1 neurons by several postischemic treatments may not be permanent (Morse and Davis, 1990; Dietrich et al., 1993; Colbourne and Corbett, 1994). Because most pharmacological studies, including those with NBQX, have used short survival times (1 week or less), the protection observed with this drug may also be transitory.
Although it has been reported that NBQX does not affect temperature (Sheardown et al., 1990), in other reports, a significant decrease in temperature after NBQX administration has been noted when temperature has been monitored for prolonged periods (Suga and Nowak, 1992; Nurse and Corbett, 1993). Using the gerbil two-vessel occlusion model, we compared the postischemic temperature profiles of animals treated with NBQX with those of vehicle controls. To determine whether mild hypothermia contributed to the beneficial effect of NBQX, we gave a third group NBQX but regulated their temperature from 1 to 24 h postischemia, so that it resembled the pattern in vehicle-treated animals. In a second experiment we simulated the NBQX temperature profile, in the absence of drug treatment, and compared the histological outcome with NBQX, at survival times of 4 and 10 days.
METHODS
Brain temperature monitoring
All experimental procedures were carried out according to the animal care guidelines of Memorial University of Newfoundland and the Canadian Council on Animal Care. Brain temperature was recorded on-line from AM-transmitter probes inserted through a guide cannula into the dorsal striatum, as described previously (Colbourne and Corbett, 1994; Nurse and Corbett, 1994). Two days before the administration of NBQX or induction of ischemia, baseline temperature was recorded for 3 h to ensure that readings were within the normal range. The sampling rate for all temperature recordings was three per minute.
Temperature effects of NBQX in normal animals
Two rodent species, Sprague–Dawley rats (n = 4) and gerbils (n = 5), were assessed for their temperature response to NBQX. Two days after initial temperature recording (see previous section), temperature probes were reinserted into the guide cannula. After baseline recording for 1 h, NBQX was administered as three separate intraperitoneal injections (3 × 30 mg/kg, i.p.), injections were spaced 15 min apart. The drug was dissolved in NaOH (0.1 M) and diluted in 5.5% glucose. Brain temperature was monitored for 24 h. NBQX (NNC 07-9202 acid) was donated by Novo-Nordisk, Copenhagen.
Cerebral ischemia
Gerbils were anesthetized with 2% halothane in 30% O2/70% N2O, and maintained at 1.5% throughout the operative procedure. The carotid arteries were isolated and occluded for 3 min at normothermia, as described previously (Nurse and Corbett, 1994). Sham-operated animals (n = 3) underwent carotid isolation without ischemia. Brain temperature continued to be monitored postischemically in unanesthetized animals for up to 30 h. During the initial 60 min of reperfusion, a 60-W lamp was used to maintain normothermia (∼37°C), if necessary. Mild postischemic hyperthermia, which occurs spontaneously, was not prevented.
Experiment I. Beginning 1 h after carotid occlusion, animals received either vehicle (5.5% glucose, n = 7) or NBQX (30 mg/kg, i.p., n = 16) at 60, 75, and 90 min postischemia. NBQX-treated animals were subdivided into two groups (n = 8 per group): NBQX and NBQX (regulated). Temperature regulation was discontinued in all groups at 60 min reperfusion, except the NBQX (regulated) group. In the NBQX (regulated) group, the brain temperature was maintained at the same level observed in the vehicle-treated group using an overhead lamp, with a variable height adjustment, until 24 h postocclusion. After the period of temperature recording (24–30 h), temperature probes were removed and the animals were returned to their home cages until 10 days postocclusion.
Experiment II. The aforementioned experiments were repeated to assess vehicle (n = 6) and NBQX (no temperature regulation, n = 9) treatments at a short survival time of 4 days. A third treatment group was also added, simulating the NBQX-induced temperature profile in the absence of drug treatment (SimTP). The NBQX-induced temperature profile was simulated manually (SimTP) for 28 h postischemia using (a) a fan placed above the cage and/or spraying room temperature water on the animal's back to lower temperature; (b) turning off the fan and/or using a 60-W lamp to elevate temperature. Animals in the SimTP group had survival times of 4 (n = 7) and 10 days (n = 10).
Histological analysis
At either 4 or 10 days postischemia, animals received an overdose of sodium pentobarbital before perfusion fixation. Brain tissue was placed in 10% phosphate-buffered formalin (pH 7.4) overnight, in situ. The following day, the brain was extracted from the skull and stored in the same fixative. Blocks were dehydrated and embedded in paraffin, sections were cut at 6 μm and subsequently stained with hematoxylin and eosin. Three sectors in dorsal CA1 (lateral, middle, and medial) were counted in each hemisphere at a level 1.7 mm posterior to bregma. The six sectors were summed to yield a total CA1 score. CA1 cell counts from six normal animals were pooled with the three sham-operated animals to provide an assessment of uninjured/normal CA1. CA1 counts were performed by D.C., who was blind to treatment status.
Statistical analysis
Total CA1 scores were analyzed with analysis of variance (ANOVA), followed by Student–Newman–Keuls post hoc comparisons.
RESULTS
When NBQX (3 × 30 mg/kg, i.p.) was given to normal animals (Fig. 1), we observed an initial drop in brain temperature of 2.0°C in gerbils and 3.5°C in rats. Three to 5 h after drug administration, temperature partially recovered but to a subnormal level, where it remained for >24 h. The changes recorded in brain temperature were also reflected in rectal temperature, which was sampled periodically in some animals (data not shown).
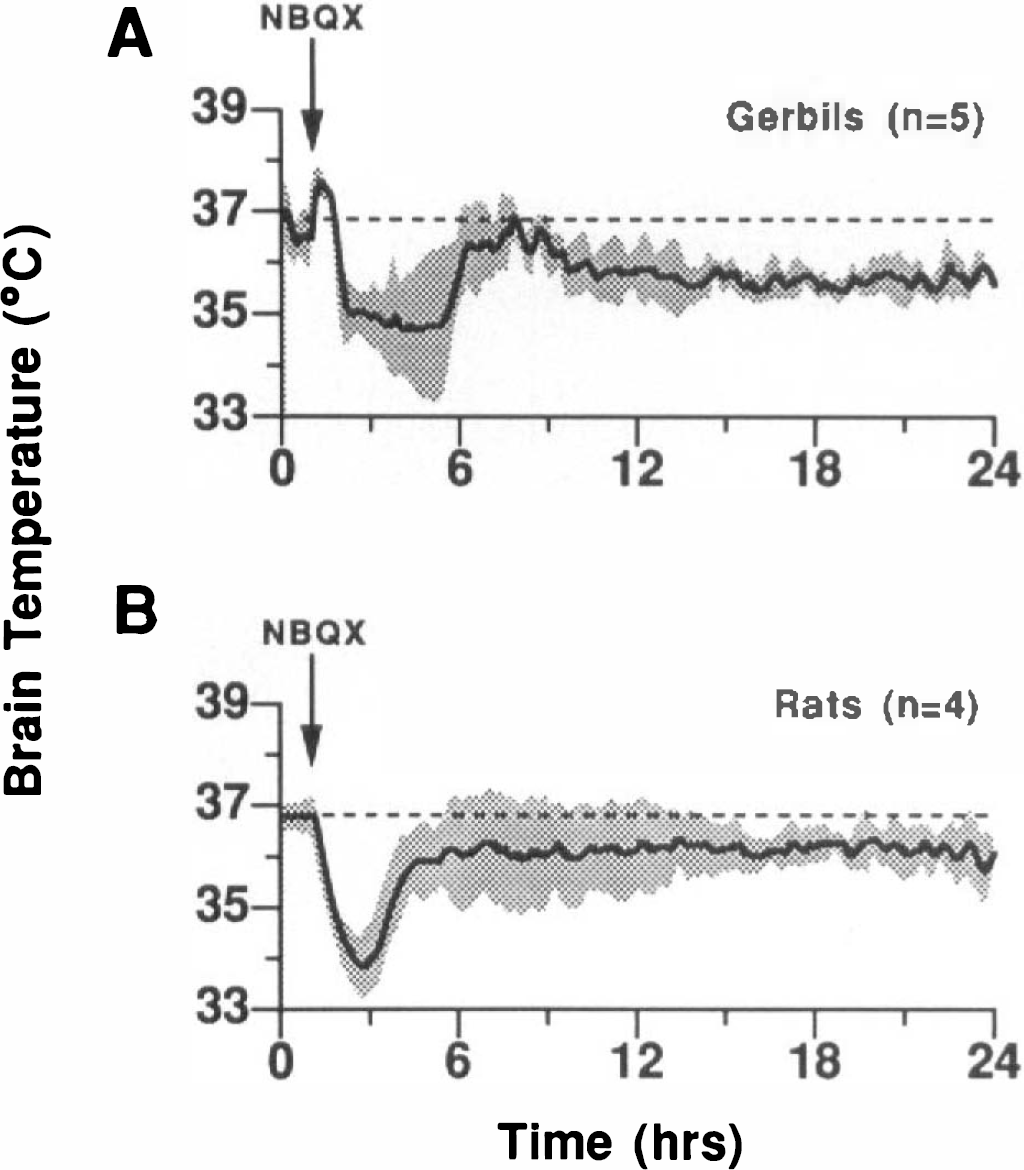
NBQX (3 × 30 mg/kg, i.p.) was administered to normal gerbils
With postischemic NBQX administration, in the unregulated condition (Fig. 2D), there was also an ∼2°C decrease in brain temperature below that observed in the vehicle-treated group (Fig. 2B), although the NBQX temperature profile was altered by ischemia (compare Fig. 1A and Fig. 2D). During ischemia, and for the initial 2 h of reperfusion, the brain temperatures of NBQX (Fig. 2D) and vehicle (Fig. 2B) groups were not different. Thereafter, the temperature of NBQX-treated animals was significantly reduced, 1.0–2.0°C, with a maximal difference (∼2.0°C) evident 2.5–3 h postischemia. Beginning 3 h postischemia, temperature gradually declined in both the NBQX- and vehicle-treated groups, leveling off ∼1.5°C below the starting point at 15 h postischemia. The slopes of the temperature profiles in the NBQX- and vehicle-treated groups were similar, with the NBQX group temperature profile being shifted 1.0–1.5°C below the vehicle group. The brain temperature in the NBQX group was still 1.0–1.5°C lower at 30 h postischemia (Fig. 2D). In an additional group treated with NBQX, we manually regulated brain temperature until 24 h postischemia (Fig. 2C) to prevent the drug-induced decrease in temperature. Interestingly, within 15 min after the termination of temperature regulation in this group, brain temperature fell 1.0°C, and remained 1.0–1.5°C below the vehicle group until the end of the recording session (Fig. 2C), which was similar to the temperature recorded in the NBQX, nonregulated group at this time (Fig. 2D).
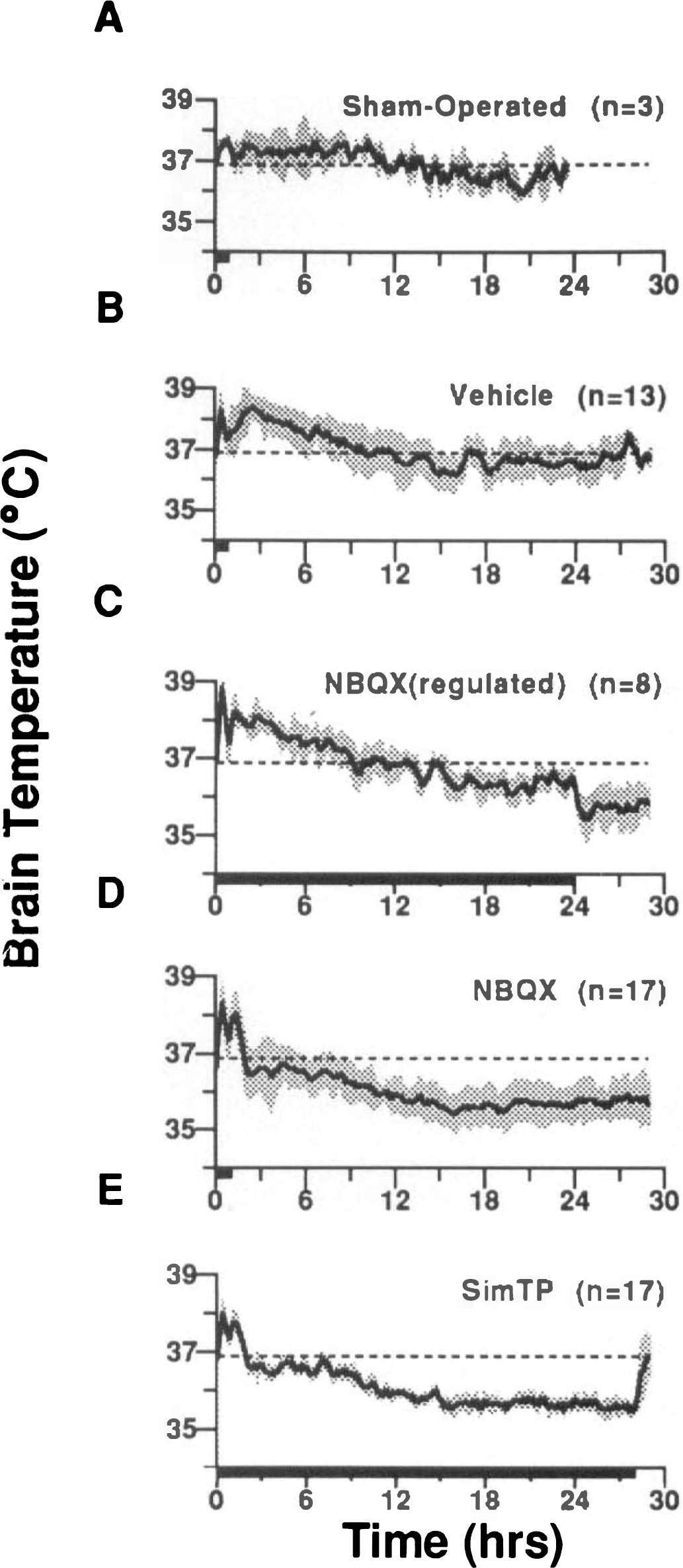
Brain temperature after ischemia/sham surgery, time 0 is the beginning of reperfusion. The profiles represent group means for each treatment, the standard deviation above and below the line is shown by the shaded area. The dashed line on each graph indicates mean normal brain temperature (36. 9°C). Vehicle (3 × 5.5% glucose, i.p.) and NBQX injections (3 × 30 mg/kg, i.p.) were given starting 60 min after ischemia, each injection separated by 15 min. The bar along the x axis indicates the duration of postischemic temperature control, which was 60 min in all groups except
Virtually all of the variance in histological outcome was attributable to these mild differences in postischemic brain temperature during the initial 24 h of reperfusion. The number of viable CA1 neurons was inversely related to the mean brain temperature from 0–24 h postischemia (Fig. 3). ANOVA of the total CA1 cell counts for normal, vehicle, NBQX, and NBQX (regulated) groups (experiment I) yielded a significant treatment effect (p < 0.0001). Post hoc comparisons of total CA1 counts revealed that the NBQX group had a significantly better histological outcome compared with both the NBQX (regulated) and vehicle groups (p < 0.01), but did have a significant degree of cell loss in comparison to normals (p < 0.01). There was no histological protection achieved with the same dose of NBQX when mild hypothermia was prevented by regulating brain temperature for 24 h to resemble the vehicle group (Fig. 3).
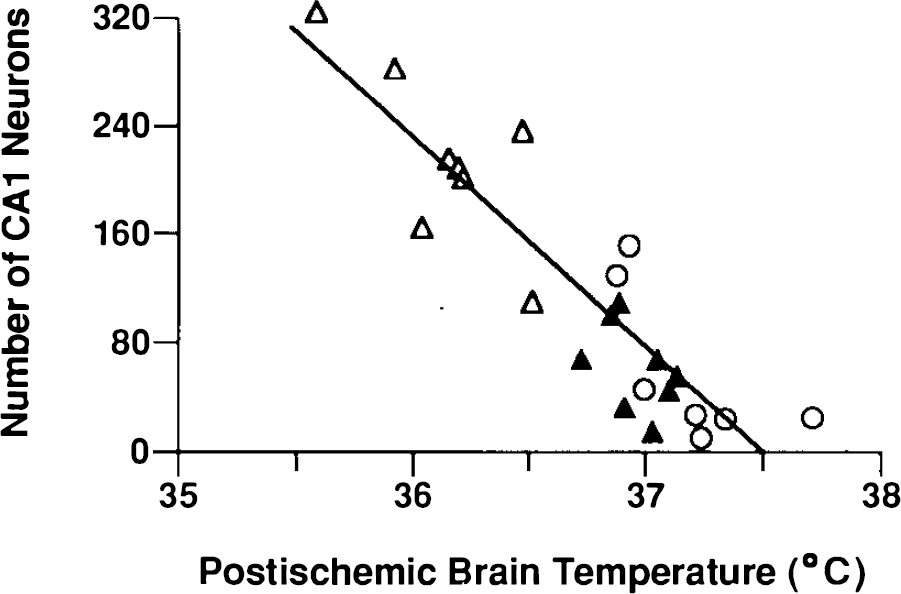
Relationship between postischemic brain temperature (averaged from 0 to 24 h postocclusion) and the number of viable CA1, neurons, r = −0.901, df = 22, p < 0.0001. Histological assessment was made 10 days after ischemia. Vehicle-treated animals (open circles), n = 7; NBQX (3 × 30 mg/kg i.p., open triangles), n = 8; NBQX with brain temperature regulated for 24 h to mimic the pattern in the vehicle group (solid triangles), n = 8.
When protection obtained with NBQX was compared with the level of protection obtained with a 28-h temperature simulation (Fig. 2E, experiment II) we found that both NBQX and the SimTP treatment produced substantial savings of CA1 neurons, which were not significantly different, when histological assessment was made 4 days postischemically (Fig. 4). Histological protection with both treatments, at 4 and 10 days postischemia, was significantly better than with vehicle [p < 0.01 in all cases, except SimTP vs. vehicle (10-day survival) p < 0.05].
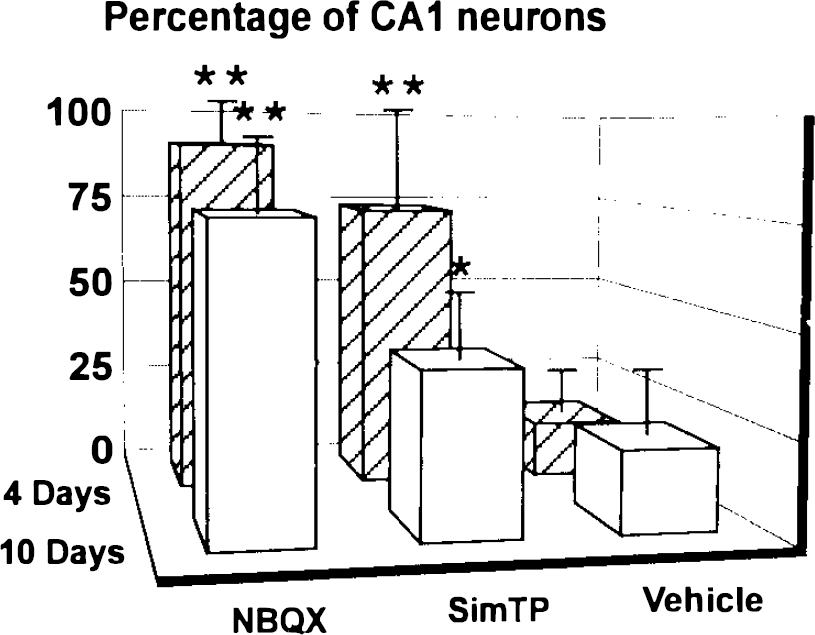
Percentage of CA-, neurons surviving 4 versus 10 days postischemia. CA1, cell counts are expressed as a percentage of normal (290.11 ± 13.72 SD; n = 9). All groups had a 3-min carotid artery occlusion with either NBQX (30 mg/kg i.p., 60, 75, and 90 min postischemia); temperature simulation to resemble the NBQX-treated group for 28 h postischemia (SimTP); or 5.5% glucose vehicle (60, 75, and 90 min postischemia). Error bars indicate the standard deviation (6–10 animals/group); *p < 0.05, **p < 0.01 in comparison to vehicle treatment.
DISCUSSION
Our results show that NBQX (3 × 30 mg/kg), a dose that has previously been reported to be beneficial against ischemic injury (Sheardown et al., 1990; Buchan et al., 1991; Diemer et al., 1992), has a mild hypothermic action that is linearly related to its neuroprotective efficacy. NBQX was ineffective against cerebral ischemia when we regulated temperature to mimic that of the vehicle group. Furthermore, simulating the temperature profile induced by NBQX is sufficient to produce histological preservation of CA1 cells. However, there was a significant decline in the protection provided by the SimTP treatment between 4 and 10 days postischemia (p < 0.01). A similar trend of decreasing efficacy was also observed with NBQX. Although not significant, this trend suggests that necrosis may progress if the survival time is lengthened to several weeks. Although it remains possible that NBQX conveys a neuroprotective effect independent of temperature, we believe that the protection provided by the SimTP treatment decayed at a faster rate than that of NBQX because the duration of mild hypothermia was shorter.
In the present study, the SimTP group was re-warmed to normal levels 28 h after ischemia; however, it is evident that NBQX-treated animals were still subnormal 30 h postischemically (Fig. 2E vs. D). Subsequently, we recorded brain temperature for extended periods (up to 8 days) in animals treated with NBQX (n = 7). NBQX induced a state of mild hypothermia that lasted for ∼4–5 days (Fig. 5), thus increasing the period of mild hypothermia by at least 68 h in comparison to the SimTP group. The long-lasting nature of this temperature effect may be due to a lingering precipitate of NBQX that is found in the intraperitoneal cavity (Le Peillet et al., 1992; Buchan et al., 1993) and internal organs, such as the gallbladder, days to weeks after i.p. administration (S. Nurse and D. Corbett, unpublished observations). This precipitate might act as a depot, providing sustained release of NBQX in high enough concentrations to produce a hypothermic action for several days. To achieve identical histological protection by simulating NBQX-induced hypothermia, the simulation period may also have to be extended to several days. This was not possible in the present study because temperature was controlled manually, and maintaining temperature at a slightly subnormal level required constant attention. However, recent findings from our laboratory (Colbourne and Corbett, 1994) have shown that 12 h of moderate hypothermia (32°C) produced substantially less protection than 24 h of hypothermia, with the shorter duration resulting in a profound decline in protective efficacy between 10 and 30-day survival times. It seems that within limits, increasing either the degree or the duration of hypothermia should improve histological outcome. Nevertheless, the present results suggest that mild postishemic hypothermia, whether drug or nondrug induced, may not translate to permanent neuroprotection even if the duration of hypothermia is prolonged (i.e., several days). In studies in which survival times have been increased from 7 days to 1 month, neuroprotection with NBQX (3 × 30 mg/kg) has been lost (Li and Buchan, 1995). The cascade of events resulting in neuronal necrosis may only be temporarily interrupted during the hypothermic period, and once temperature is normalized these processes may resume, albeit at a slower rate. In the present study, and others (Sheardown et al., 1990), in which NBQX has been assessed after survival times of only 4 days, histological protection is often indistinguishable from normal cell counts/ratings. According to the findings presented here, the animals would have still been mildly hypothermic at this time, which could account for the near total histological preservation.
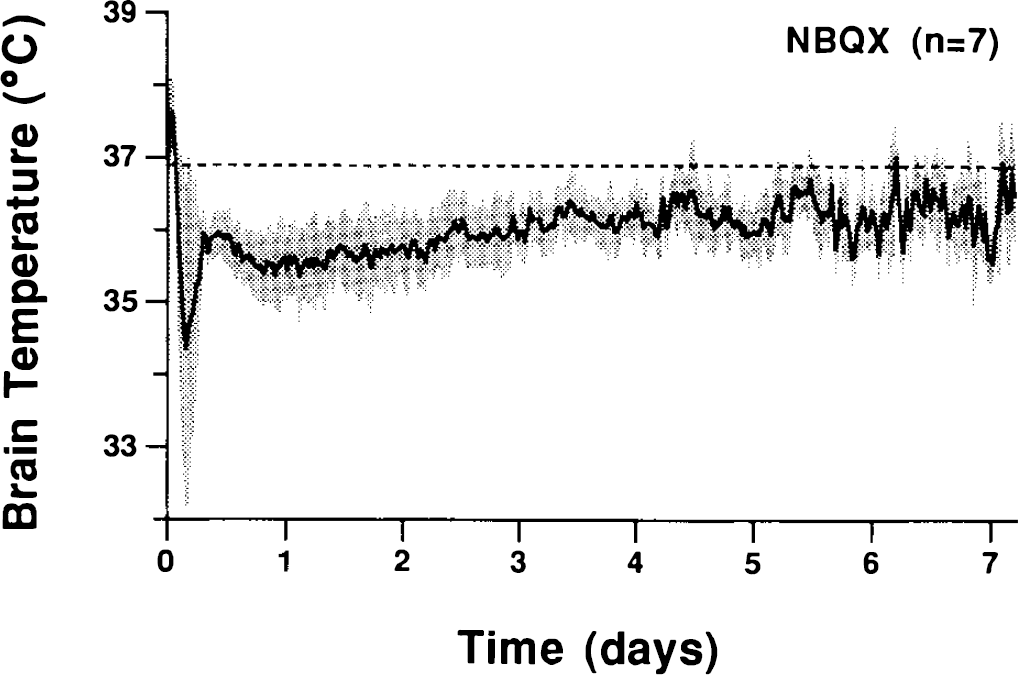
Brain temperature recorded postischemically for 1 week after NBQX treatment (30 mg/kg, i.p.) administered at 60, 75, and 90 min postischemia (time 0 is the start of reperfusion). The shaded area represents a standard deviation above and below the mean. Temperature data have been averaged over 30-min intervals. The dashed line indicates mean normal brain temperature (36.9°C).
Although the majority of the previous studies using NBQX have not regulated temperature during the postischemic period, a study by Buchan et al. (1991) reported significant histological savings at 7 days when body temperature was regulated postischemically for 6–8 h. However, as we have shown (Fig. 2C), the temperature of NBQX-treated animals drops almost immediately even when temperature regulation is terminated as late as 24 h. It is also noteworthy that animals treated with NBQX demonstrate hypothermia even when they recover from the mild sedating effects of the drug and begin to show normal ambulation. Recovery of locomotion is often used as a marker of an animal's ability to thermoregulate after ischemia and a point at which temperature control is typically stopped, this is clearly an incorrect assumption. Because NBQX administration can be delayed for 6–12 h (Li and Buchan, 1993; Nurse and Corbett, 1993) and sometimes 24 h (Sheardown et al., 1993; but see Li and Buchan, 1993) postischemia and still provide neuroprotection, then postischemic temperature regulation for <24 h is likely to be insufficient if neuronal protection is due to hypothermia. The severity of the ischemia will probably determine the length of time that is necessary to regulate postischemic temperature and prevent hypothermia from confounding histological outcome. For example, after 5 min of ischemia, which produces >95% CA1 cell loss in our model, we have found NBQX treatment to be ineffective if administration is delayed until 6 h after reperfusipn (Nurse and Corbett, 1993). Therefore, with severe ischemia, if NBQX is given within the first few hours of reperfusion then a 6–8-h period of temperature regulation would probably suffice. However, after 3 min of ischemia, which produces ∼85% CA1 neuronal loss in our model, treatment can be delayed for at least 6 h and provide histological protection (data not shown). However, the protection was correlated with the level of hypothermia induced, and no histological protection was found after the 6-h delayed administration if temperature was regulated from 6 to 24 h postischemia (Nurse and Corbett, 1993). In models where CA1 cell loss is ∼80% or less (Sheardown et al., 1993), temperature regulation may need to be continued beyond 24 h to prevent a confounding effect of hypothermia.
This study also helps to reconcile some of the discrepancies between in vivo and cell culture models of ischemia and glutamate toxicity. Many in vivo studies comparing the efficacy of NMDA and AMPA glutamate antagonists in global ischemia have found AMPA antagonists to offer considerably greater levels of neuroprotection (Nellgård and Wieloch, 1992; Buchan et al., 1993; Sheardown et al., 1993; Lippert et al., 1994). In contrast, cell culture studies have either found no neuroprotective effects with AMPA antagonists alone (Kaku et al., 1991), or in cases in which AMPA antagonists were effective the protection was not as great as that provided by NMDA antagonists (Lippert et al., 1994). However, both in vitro (Shuaib et al., 1993; Bruno et al., 1994) and in vivo (Busto et al., 1987; Dietrich et al., 1993; Coimbra and Wieloch, 1994; Colbourne and Corbett, 1994; Nurse and Corbett, 1994) studies have demonstrated dramatic protection with hypothermia. It is important to emphasize that the level of postischemic hypothermia induced by NBQX is milder than that in previous studies (Busto et al., 1989; Dietrich et al., 1993; Coimbra and Wieloch, 1994; Colbourne and Corbett, 1994, 1995) using deliberate hypothermic manipulations (e.g., 32–34°C). These results demonstrate the necessity of careful postischemic temperature assessment in pharmacological studies of cerebral ischemia because differences of only 1.0–2.0°C, if prolonged, can significantly affect outcome. Furthermore, this study raises important questions about the wisdom of conducting costly clinical trials based on current drug studies that are likely confounded by similar temperature effects and short survival times.
Footnotes
Acknowledgment:
This study was supported by the Medical Research Council of Canada, grant MT-10196 awarded to D.C. We thank Dr. John Evans, Dr. Richard Neuman, Dr. Carolyn Harley, Suzanne Evans, Fred Colbourne, and Mark Hawryluk for their helpful comments on the manuscript and Suzanne Evans and Kathleen McKay for their expert assistance.