
Abstract
The consequences of an unilateral electrolytic entorhinal lesion on the functional activity in all major anatomically defined brain regions were evaluated in the rat. The 14C-2-deoxyglucose method served as a tool to quantify alterations of local cerebral glucose utilization (LCGU) ipsilateral and contralateral to the lesion at 4 days, 2 weeks, or 3 months after stereotaxic surgery. Apart from a few minor increases in the contralateral hemisphere, the predominant pattern consisted of reductions in the range of 10–40% in the ipsilateral hemisphere. Ipsilaterally, in extrahippocampal areas, LCGU had regained control levels at 2 weeks postlesion in contrast to hippocampal regions, where reductions were more pronounced than in other brain areas and partially persisted for up to 3 months. Interestingly, the termination zones of entorhinal fibers in the dentate gyrus did not regain control levels within 3 months. We conclude from the data that functional recovery of denervated primary target areas does not occur within 3 months after entorhinal lesions and that altered functional activity may be found beyond the primary target areas predominantly during the acute recovery period after the lesion. The data suggest that sprouting fibers do not reestablish a fully functional neuronal network during the recovery period.
Entorhinal lesions have been employed in numerous studies seeking to neurochemically characterize the processes governing the sprouting of new fibers in denervated hippocampal regions. It is well known that de-entorhination leads to a loss of ∼80–90% of synapses in the outer two-thirds of the molecular layer of the dentate gyrus, while the inner one-third remains largely unaffected, receiving its input mainly from commissural and associational fibers (Matthews et al., 1976a). Though only driven by a denervation of <5%, the contralateral molecular layer also initially exhibits synapse loss of 15% and subsequent replacement to control levels within months (Hoff et al., 1981). Invariably, following creation of the lesions, massive sprouting occurs ipsilaterally in the denervated zone. Synaptic density gradually increases after the lesion is made due to the invasion of newly formed fibers, notably commissural/associational fibers from the inner molecular layer, septal afferents, and the crossed temporo-ammonic tract, the latter two both terminating in the denervated outer molecular layer (Lynch et al., 1974; Matthews et al., 1976a,
b
; Amaral et al., 1980). At first glance, one would assume this reactive synaptogenesis to be an expression of regeneration within the denervated area. It is, however, questionable if these morphological phenomena connote functional recovery as well or only reflect a neuronal circuitry in continued disarray. Recent findings strongly suggest that restoration of the pre-lesion neuronal network is not achieved. Nitsch and Frotscher (1993) showed that apart from early dendritic swellings, the number of parvalbumin-immunoreactive (PV-ir) dendrites in the dentate gyrus were persistently reduced for up to 1 year after the entorhinal ablation, while PV-ir cells were not reduced. At least part of this transneuronal degeneration appears to be mediated by N-methyl-
MATERIAL AND METHODS
Animals
Experimental procedures were carried out with adult male Wistar rats weighing 300–330 g (Winkelmann, Borchen, Germany). Rats were housed at 22 ± 2°C under controlled environmental conditions and had free access to tap water and a standard diet (Altromin, Lage, Germany).
Entorhinal lesion
Lesions of the entorhinal cortex were created at five stereotaxic coordinates by passing an anodal direct current (DC) increasing at increments of 5 mA/s from 5 to 30 mA through an unipolar stainless steel electrode with a tip diameter of 0.8 mm (Olton, 1975). Coagulation was performed at 30 mA. Duration of current was 8 s. This procedure has proven to be superior to applying the maximum current of 30 mA from the beginning in that the lesion size covers the entire entorhinal cortex. Had we applied 30 mA from the onset, tissue would have been coagulated only within a small demarcated area around the tip of the electrode and the current quickly fallen to zero level. Stereotaxic coordinates were caudally at an angle of 25° to the vertical plane: AP 2.0 mm, lateral 4.2 mm, and DV 1.6 and 2.2 mm, and AP 2.0 mm, lateral 5.2 mm, and DV 1.6 and 2.2 mm; and rostrally at angles of 25° to the vertical plane and 6° to midline: AP 3.7 mm, lateral 6.4 mm, and DV 0.8 mm (Fig. 1). Animals survived for 4 days, 2 weeks, or 3 months and subsequently underwent the 14C-2-deoxyglucose procedure. Controls were sham-operated and received the same treatment except for passing a current through the electrode.
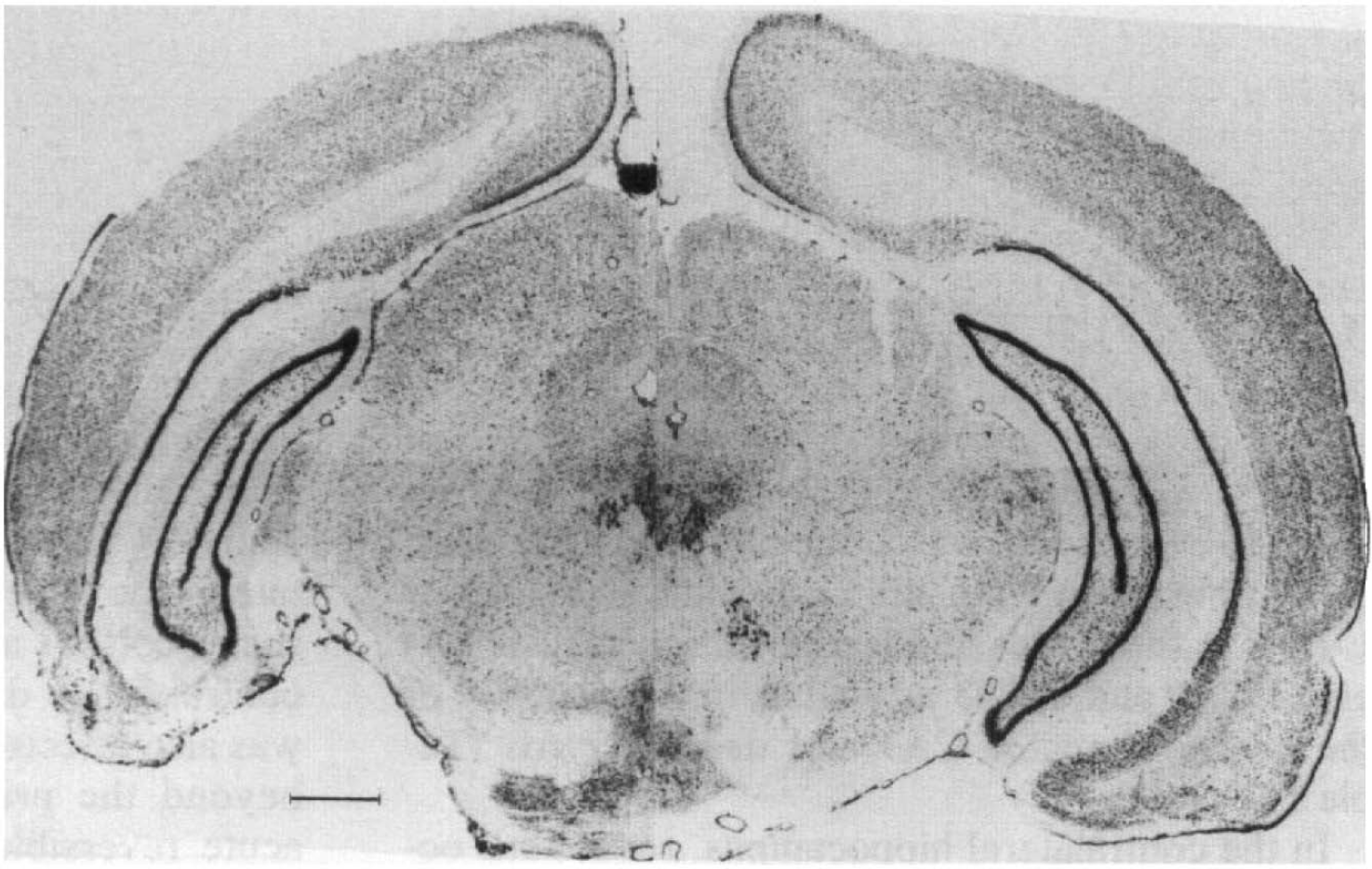
Nissl-stained coronal section of rat brain illustrating unilateral electrolytic lesion of entorhinal cortex. Section taken at 2.7 mm anterior to interaural plane. Arrowheads denote boundaries of entorhinal cortex, star denotes lesioned area. Bar = 1 mm.
Determination of local cerebral glucose utilization (LCGU)
Procedures followed the guidelines given by Sokoloff et al. (1977). In brief, rats were anesthetized with 1% halothane in a 70:30 (vol/vol) mixture of nitrous oxide and oxygen. The femoral artery and vein were cannulated and lidocaine gel applied to the wound before it was closed. Then, animals were lightly restrained with plaster casts covering the hindlimbs and lower abdomen, leaving fore-limbs, and thorax free. Temperature was held at 37°C. After a recovery period of 2h and a check of physiological variables, 80 μCi/kg 14C-2-deoxyglucose (Biotrend, Köln, Germany) dissolved in physiological saline was injected via the femoral vein within 20 s. During the ensuing 45 min, 16 timed arterial blood samples were drawn from the arterial catheter, immediately centrifugea, and assayed for radioactivity and plasma glucose. After 45 min, rats received a lethal dose of pentobarbital and were decapitated. Brains were rapidly dissected out and frozen in isopentane chilled to −50°C. Cryostat sections of 20 μm thickness were cut, thaw-mounted on coverslips, glued to card board, and exposed to Osray M3 film (Agfa-Gevaert, Leverkusen, Germany) together with precalibrated 14C-methylmethacrylate standards for 14 days.
Image analysis
Image analysis was performed with a VIDAS image analyser (Kontron, Eching, Germany). Autoradiograms were digitized with a resolution of 14 × 14 μm per pixel. Images were then printed as hard copies that were used for delineations of anatomical structures by superimposing the prints with adjacent Nissl- or acetylcholinester-asestained sections using a drawing tube attached to a microscope. Additional sections were also stained for myelin according to a modified Gallyas method (Zilles, 1985). Grey values were transformed to glucose utilization by using the operational equation for changing arterial plasma levels (Savaki et al., 1980). Finally, regional glucose utilization was measured by superimposing the delineated hard copies with the topographical data array of glucose utilization values stored in the computer and by selecting the region of interest according to the anatomical boundaries marked previously. For each structure, 10–12 autoradiograms were measured. Due to the small size of the respective areas, we did not divide the dentate gyrus molecular layer into a preserved inner one-third and a denervated outer two-thirds, since tracing of the boundaries became too variable.
RESULTS
Histologically, staining for acetylcholinesterase gradually increased after the lesion in the outer two-thirds of the dentate molecular layer and the lacunosum-molecular layer of the CA1 on the lesioned side (Figs. 2, 3). Along with this, increased staining for myelin was clearly diminished within the same areas (Fig. 2).
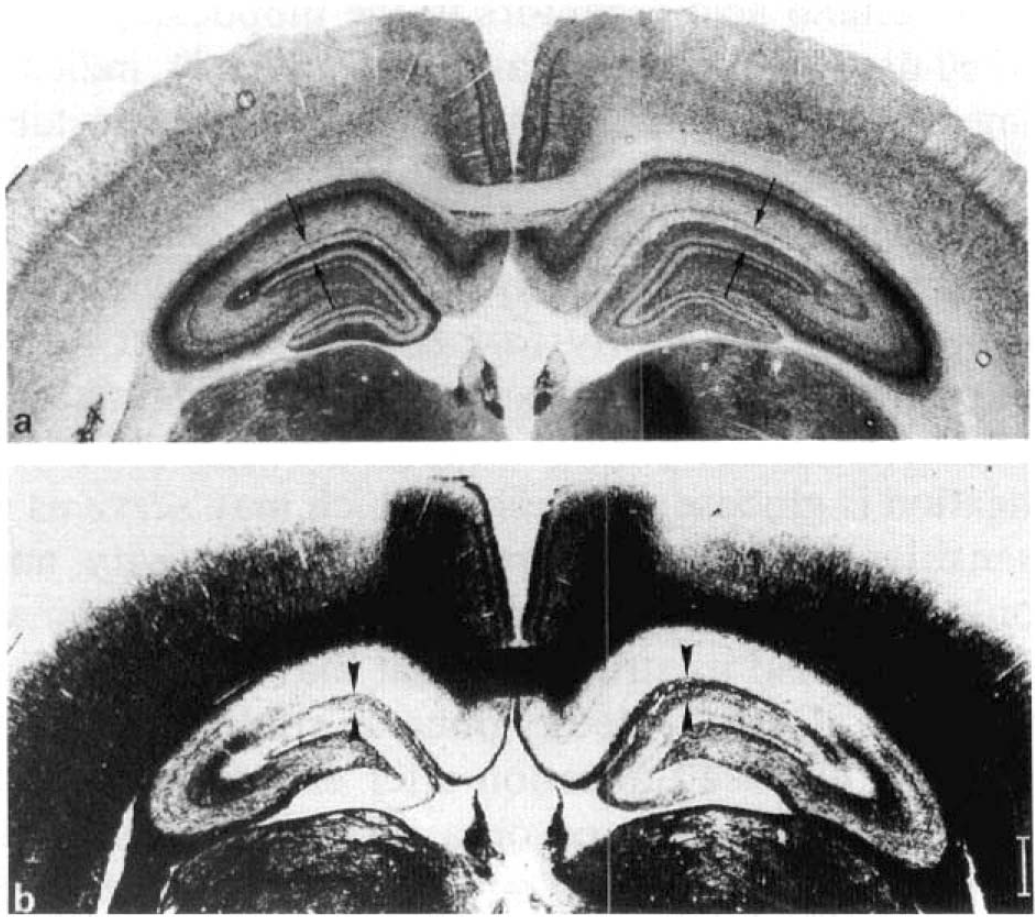
Coronal sections of rat brain 3 months after unilateral lesion of the entorhinal cortex. Lesioned side includes left hemispheres in
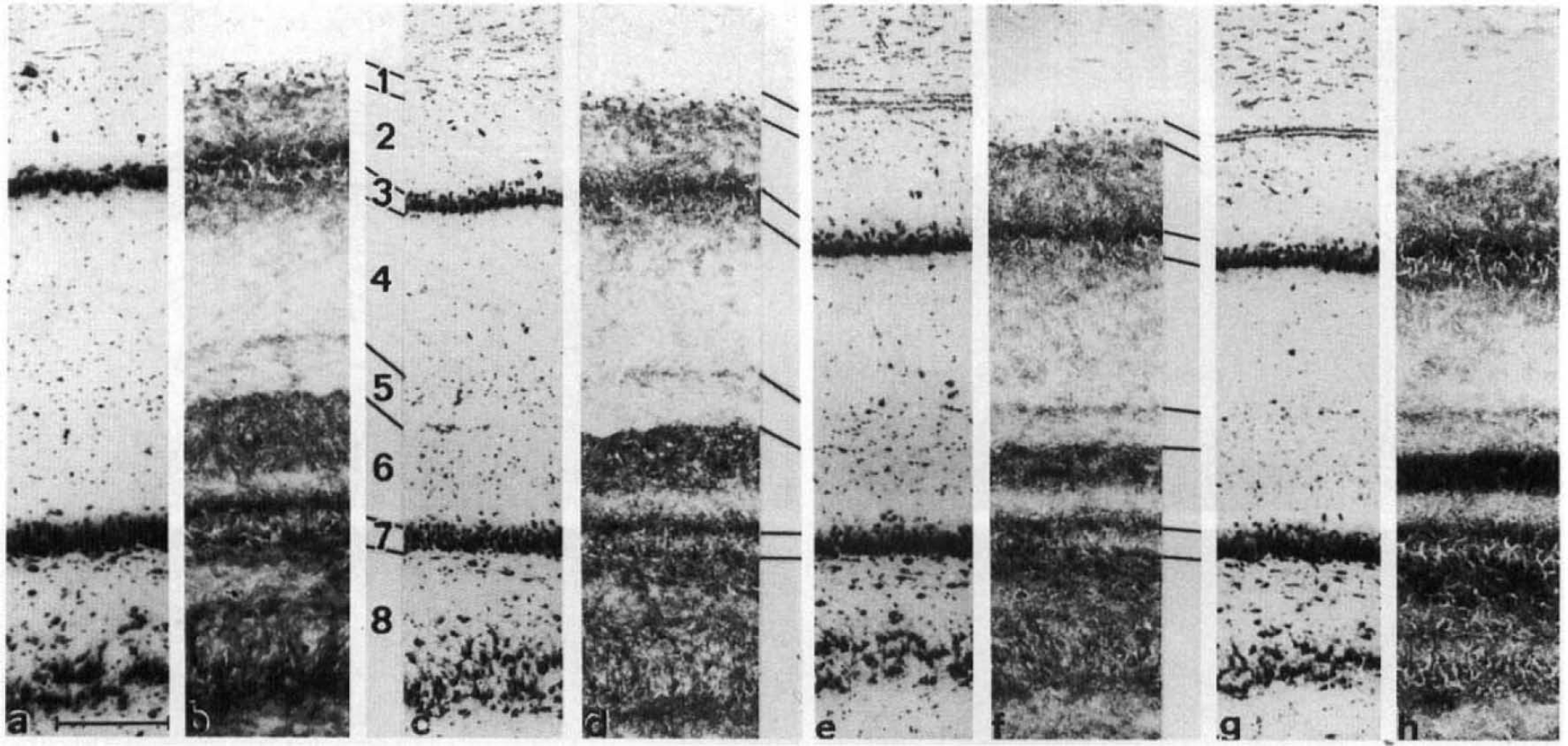
High-power photomicrographs of consecutive coronal rat dorsal CA1 and upper dentate gyrus taken at ∼5.2 mm anterior to the interaural plane stained for perikarya
Significant alterations of LCGU occurred primarily ipsilateral to the lesion and were particularly pronounced in the hippocampus (Fig. 4). LCGU in the hippocampus was 15–39% lower than in sham-operated controls. At 4 days postlesion, all hippocampal layers and sectors were affected except for the alveus in CA1 and CA3. The pattern of reductions of LCGU was almost unchanged 2 weeks after the entorhinal lesion. Only the oriens layer of CA1 and the pyramidal layer of CA2 showed a significant return to control levels. At 3 months, reductions were less pronounced compared to earlier time points, but there were still significant decreases of LCGU in the alveus and lacunosum-molecular layers of CA1 and CA2/3 as well as in the majority of the layers in sector CA3 and dentate gyrus (Table 1).
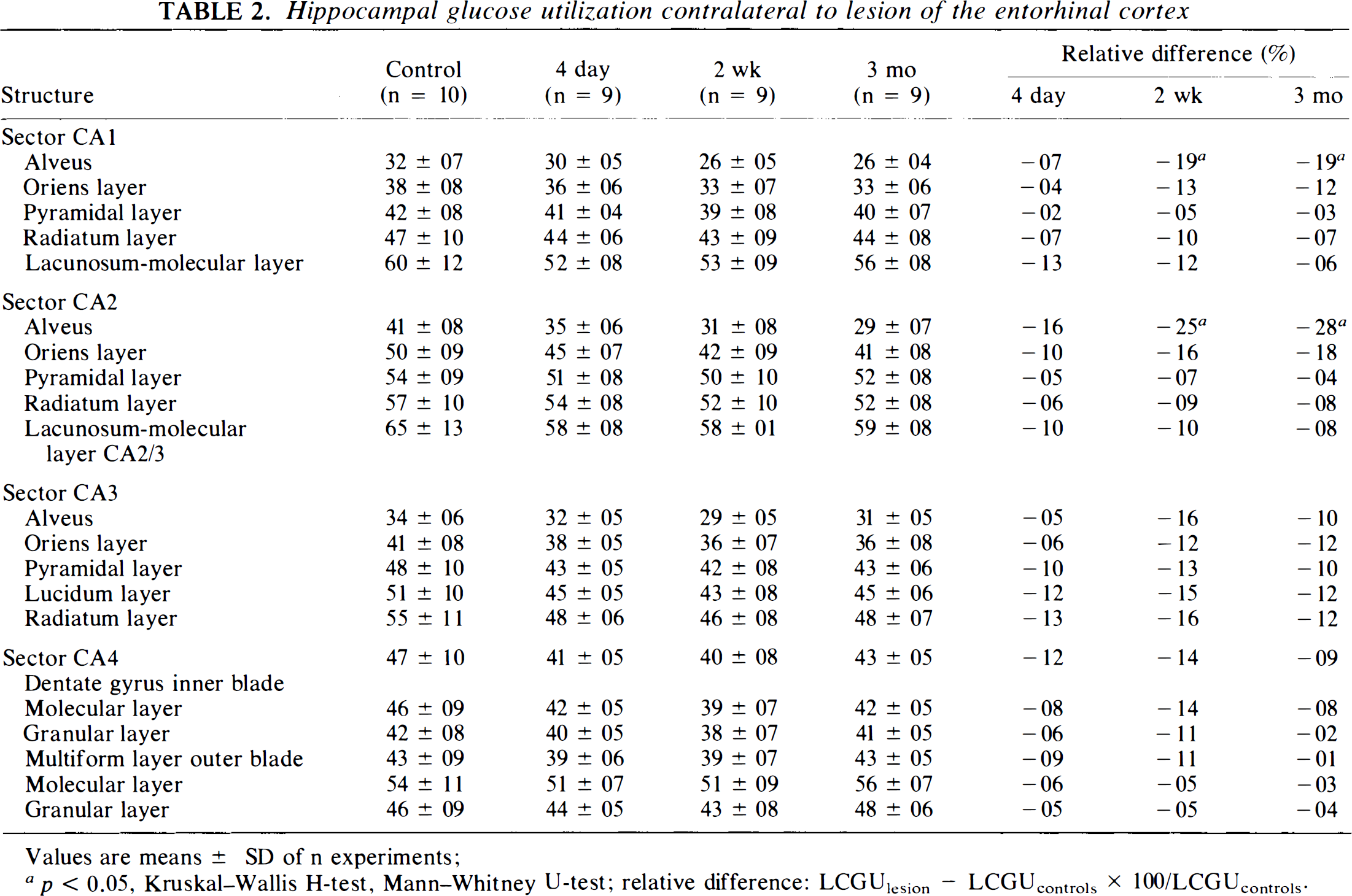
Hippocampal glucose utilization ipsilateral to lesion of the entorhinal cortex
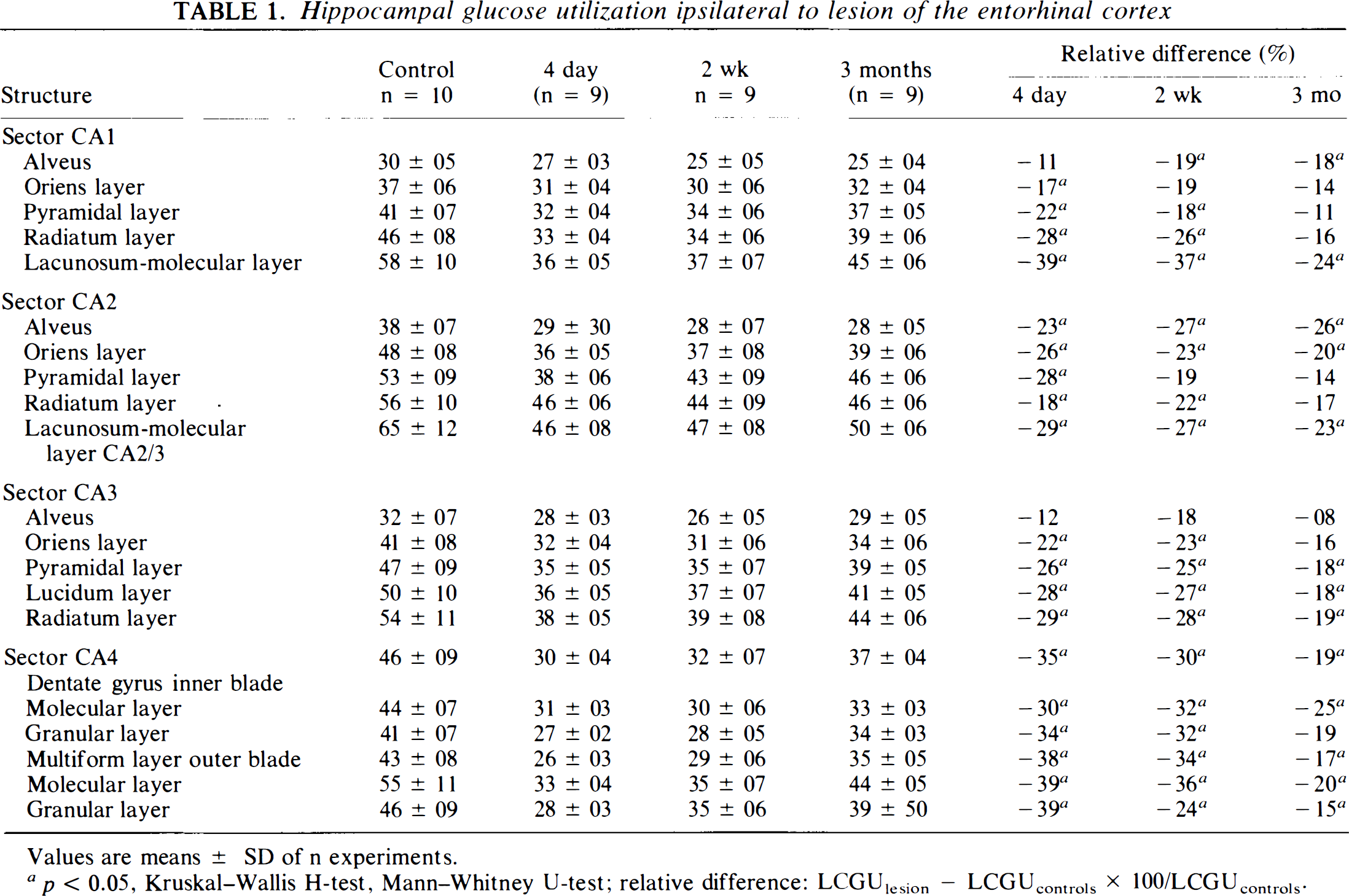
Values are means ± SD of n experiments.
p < 0.05, Kruskal-Wallis H-test, Mann–Whitney U-test; relative difference: LCGUlesion − LCGUcontrols × 100/LCGUcontrols.
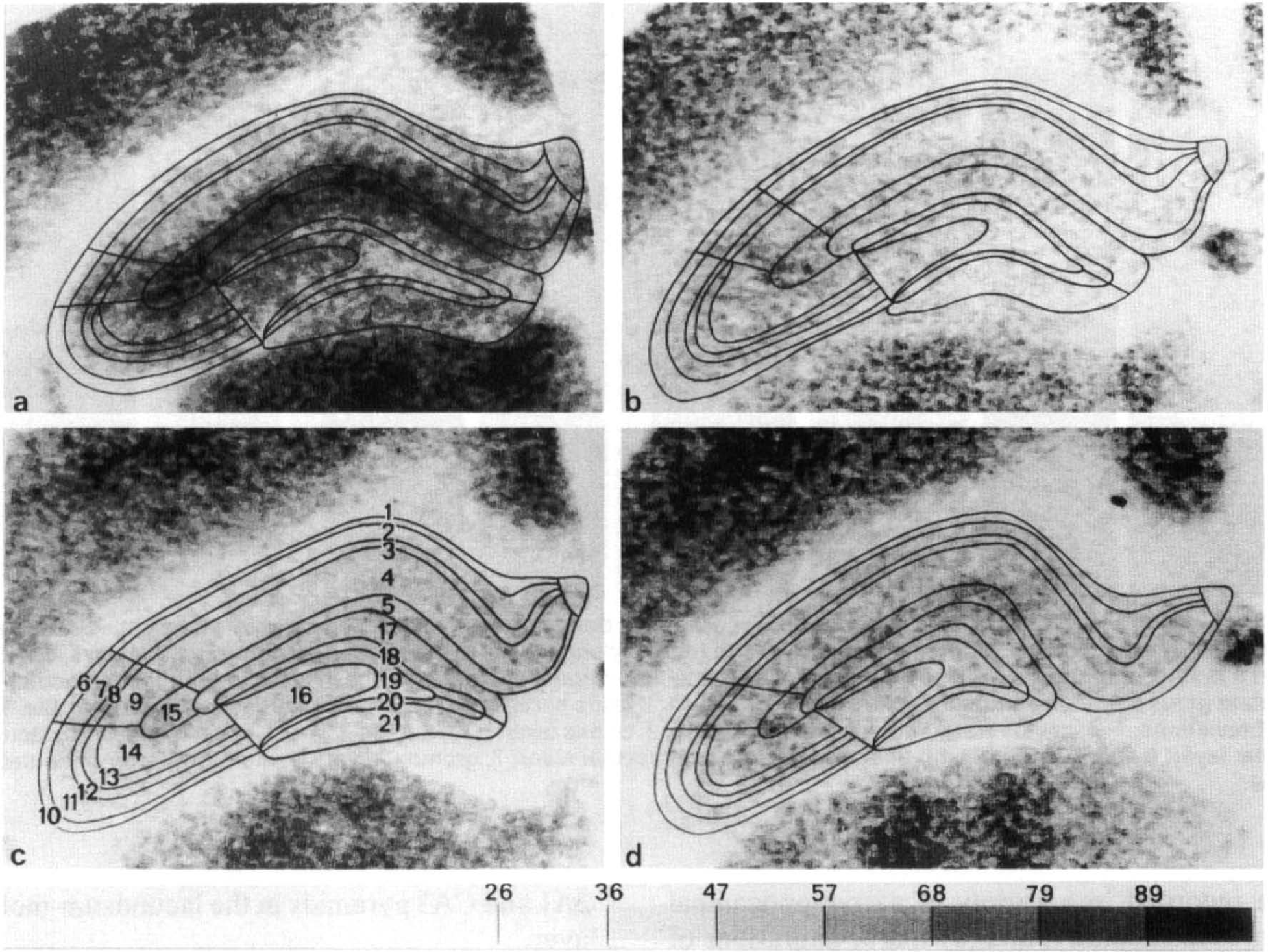
Coronal section of rat brain dorsal hippocampus at the level of ∼5.2 mm anterior to the interaural plane. 14C-2-deoxyglucose autoradiograms transformed to LCGU given in μmol/100g/min. Different ranges of LCGU are plotted in different print modes according to the scale given at the bottom. In the LCGU prints, areal and laminar boundaries are traced by superimposing the prints on the adjacent Nissl- and acetylcholinesterase-stained sections,
In the contralateral hippocampus, reductions occurred 2 weeks and 3 months after the lesion and were limited to the alveus in sector CA1 and CA2 (Table 2). Reductions were also found in extrahippocampal structures, predominantly ipsilateral to the lesioned entorhinal cortex. However, they were relatively short-lived, persisted only up to 4 days postlesion, and had vanished at 2 weeks and 3 months postlesion (Table 3). Contralaterally, slight increases occurred in area 1 of temporal cortex, and in the prepiriform cortex and the inferior colliculus 3 months postlesion. The prepiriform cortex showed also a significant decrease 4 days postlesion (Table 4).
Hippocampal glucose utilization contralateral to lesion of the entorhinal cortex
Values are means ± SD of n experiments;
p < 0.05, Kruskal-Wallis H-test, Mann–Whitney U-test; relative difference: LCGUlesion − LCGUcontrols × 100/LCGUcontrols.
Local cerebral glucose utilization ipsilateral to electrolytic lesions of rat entorhinal cortex
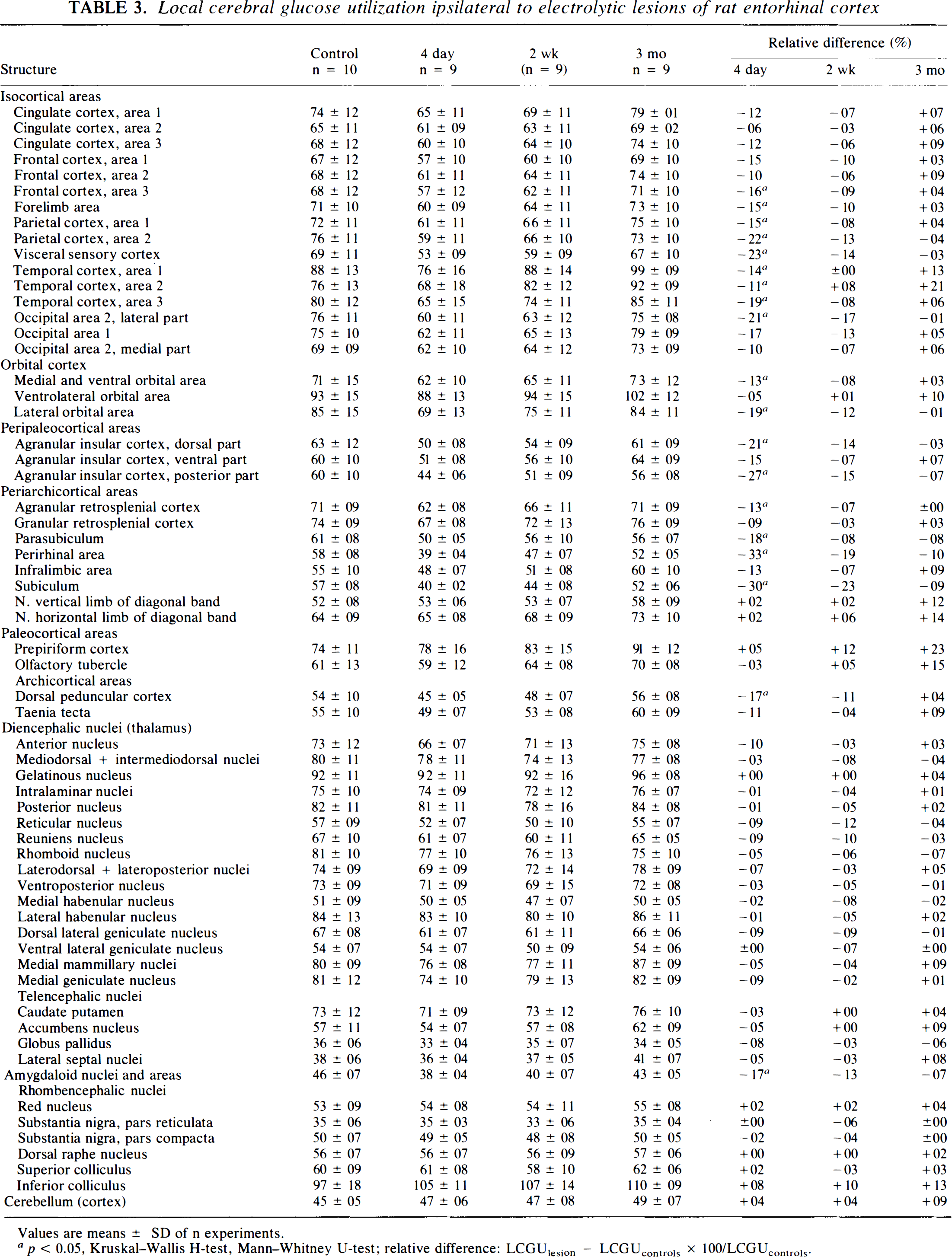
Values are means ± SD of n experiments.
p < 0.05, Kruskal-Wallis H-test, Mann–Whitney U-test; relative difference: LCGUlesion — LCGUconlrols × 100/LCGUcomrols.
Local cerebral glucose utilization contralateral to electrolytic lesions of rat entorhinal cortex
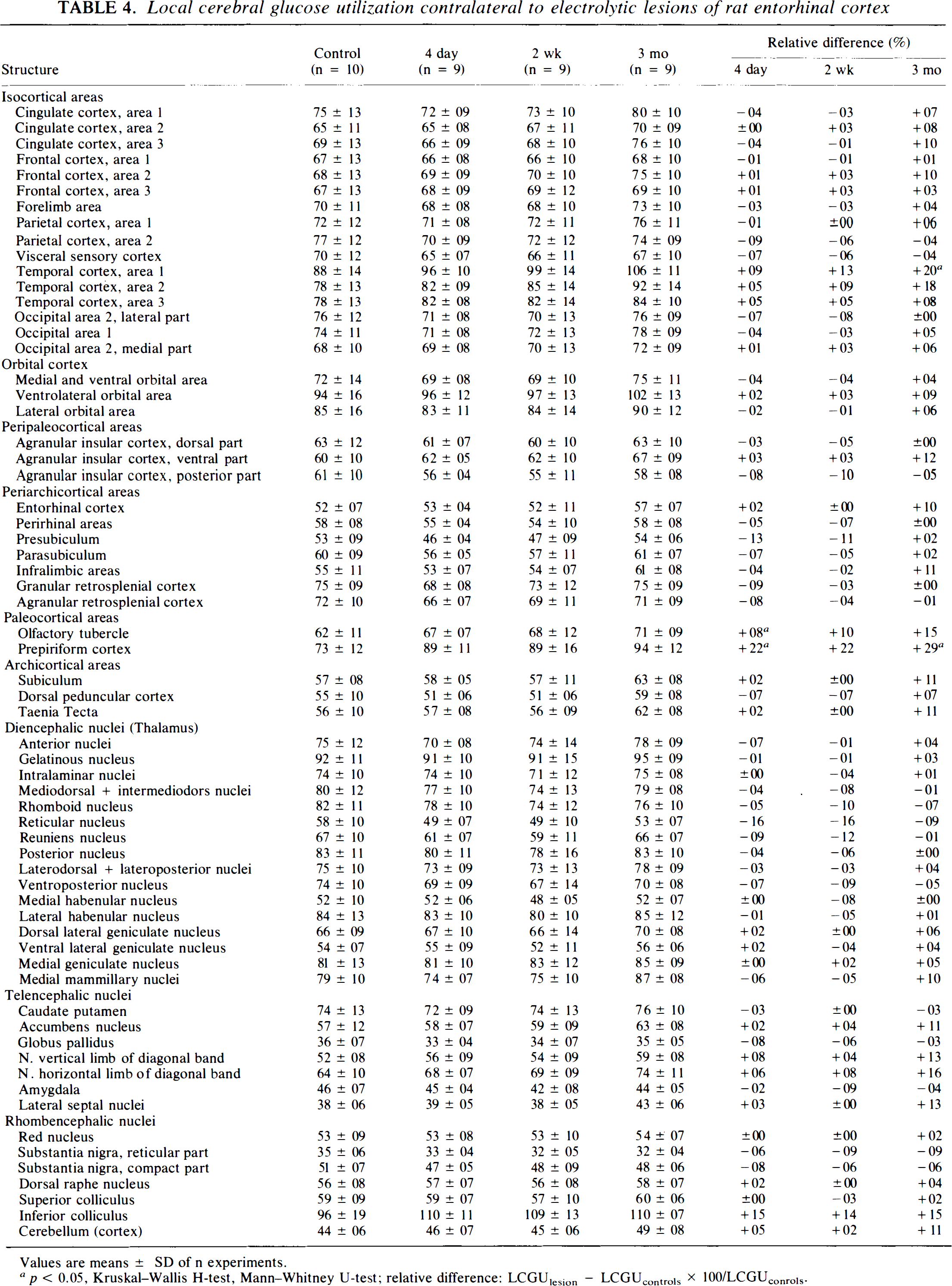
Values are means ± SD of n experiments.
p < 0.05, Kruskal-Wallis H-test, Mann–Whitney U-test; relative difference: LCGUlesion — LCGUcontrols × 100/LCGUconrols.
DISCUSSION
The present work describes alterations of functional activity after unilateral lesion of the entorhinal cortex quantified by measurements of local cerebral glucose utilization. The salient points of the study are (a) within the areas of the ipsilateral dentate gyrus denervated by the ablation of the entorhinal cortex, a recovery of functional activity to control levels did not occur, (b) glucose utilization was also affected in ipsilateral hippocampal regions beyond the primary entorhinal target areas, (c) acute reversible reductions of glucose utilization can be recorded in a variety of extrahippocampal structures ipsilaterally, and (d) sparse alterations of glucose utilization occurred in the contralateral hemisphere.
The hippocampus has been a prime object for studying the influences on an intrinsic neuronal network of afferent fibers, mainly due to its lamellar organization, which receives synaptic contacts in a precisely defined manner. The well-known localization of the links of this neuronal network provides the basis for selective lesions of this circuitry to assess the significance of the lesioned link for the remainder of the neuronal network lying downstream to the lesion.
The entorhinal cortex provides the bulk of excitatory input to the hippocampus via the perforant path. Strictly speaking, this tract branches off into the temporodentate and the temporoammonic branches. The temporodentate branch, originating mainly from layer II neurons in the medial and lateral entorhinal areas, crosses the hippocampal fissure synapsing on dendrites of granular cells of the dentate gyrus within the outer two-thirds of the molecular layer. The granular cells, in turn, give rise to the mossy fibers contacting the CA3 pyramidal cells that finally contact the CA1 pyramids via the Schaffer collaterals. Apart from this trisynaptic pathway, the temporoammonic tract emanates from layer III of the entorhinal cortex, stays within the cornu am-monis, and converges directly on the dendrites of CA1 and CA3 pyramids in the lacunosum-molecular layer.
It has been shown that this circuit responds characteristically to a removal of the entorhinal input. Once the entorhinal cortex is lesioned, the outer two-thirds of the molecular layer of the dentate gyrus are denervated, while the innervation of the inner portion of the molecular layer by commissural and associational fibers remains intact. Loss of synapses occurs rapidly (within 2–4 days), but is quickly replaced by formation of new synapses starting at day 8 after the lesion and peaking at days 12–14 postlesion (Matthews et al., 1976a; Hoff et al., 1982). Steward and Vinsant (1983) calculated that 1 month after the lesion, 70% of synaptic contacts are in place again, with a continued minute increase up to 6 months postlesion. Commissural/associational fibers extend their terminal fields into the denervated zone, a region in which they do not normally synapse (Cotman and Nieto-Sampiedro, 1984), and gradually occupy the inner one-half of the molecular layer. A second source of reinnervation is comprised of fibers from the septum, occupying the vacated terminal field in the outer two-thirds of the molecular layer and leading to a significant increase of acetylcholinesterase in this area (Lynch et al., 1972). The marked increase of acetylcholinesterase staining is taken as a landmark for successful entorhinal ablations, and our staining pattern after the lesion is in line with this requirement, as is the reduced staining for myelin, which indicates the loss of myelinated axons within the outer molecular layer.
With these dynamic changes in mind and considering that the postsynaptic cell body may be reasonably expected to be the site for neuronal energy consumption, one would forecast a biphasic time course of glucose utilization after de-entorhination: a gradual initial decline of LCGU in the denervated area reflecting the ongoing synaptic degradation and associated loss of excitatory input, and a subsequent increase due to the replacement of synapses as a consequence of ingrowing fibers either from the inner molecular layer of the dentate gyrus or from the septum reestablishing the network and restoring functional activity. However, our data do not support such a scenario. The persistent decrease of LCGU in major regions in the primary target areas of the perforant path within the hippocampus shows that functional activity does not regain control levels within 3 months after the lesion, but remains on a significantly decreased level that is below normal control values. In fact, the decrease may have been even more pronounced in the denervated outer two-thirds, since our measurements were obtained from the entire molecular layer of the dentate gyrus. As already pointed out above, a delineation based on the deafferented area in the dentate gyrus would have introduced errors. Interestingly, the following relay stations of the trisynaptic pathway located in the CA3 and the CA1 sectors are also affected and depict a chronic depression of glucose metabolism. Without considering the sprouting response as normally seen with entorhinal cortex ablation, this result could be expected on theoretical grounds and be in line with the idea that it is primarily the excitatory input from the entorhinal cortex that is driving the activity of the trisynaptic pathway and, thereby, determining the resting hippocampal glucose utilization that is needed for fueling the energy demand of the tissue. In keeping with this notion, a previous study reported reductions of glucose utilization of comparable magnitude in the termination zone of the entorhinal projection as well as in the interconnected hippocampal areas 4 days after aspiration of the entorhinal cortex when synaptic replacement can still be neglected (Jørgensen and Wright, 1988). On the other hand, however, there is a discrepancy with one report on unchanged cerebral glucose utilization 14 days after entorhinal lesion in the rat (Kurumaji and McCulloch, 1990). It may, however, be misleading to compare these data directly, since those workers used a single injection of ibotenic acid to lesion the caudal aspects of the entorhinal cortex, while for the present study, an electrolytic lesion at five different coordinates was used, producing a more complete entorhinal lesion that may well correspond in size to the aspirative lesion induced by Jørgensen and Wright (1988), showing an acute effect comparable to the present data. As pointed out, any 2-deoxyglucose investigation covering a longer time course will reflect the various cellular processes involved in the degeneration and regeneration over time. Due to its nature, the 2-deoxyglucose method provides data only on the total sum of energy consumed in a given tissue area irrespective of the cellular components contained therein. For this reason, metabolism of neurons as well as of glial cells may contribute to the final level of glucose utilization. Glial response to lesions of the entorhinal cortex has been comprehensively documented. In the denervated areas, microglia increase within the first day after the entorhinal lesion, paralleled by rising levels of interleukin-1, followed by hypertrophy and proliferation of glial fibrillary acidic protein (GFAP)-positive astrocytes starting 5 days postlesion (Rose et al., 1976; Gall et al., 1979; Gage et al., 1988; Fagan and Gage, 1990). IL-1 returns to control levels at day 5 postlesion and GFAP normalizes on the protein as well as on the message levels between 10 and 30 days postlesion (Fagan and Gage, 1990; Steward et al., 1990). As documented previously, glial cells may show a high metabolic demand and, hence, high glucose metabolism (Benson et al., 1985; Duncan et al., 1987; Komatsumoto et al., 1989). One may, therefore, reasonably assume that without the “glial share” of the functional activity measured, the drop in metabolic activity in the entorhinal target areas should be even more marked. In line with this depression of functional activity, electrophysiological experiments show that firing of dentate granule cells is significantly reduced after an entorhinal ablation at least for days after the lesion (Reeves and Steward, 1988). Moreover, analysis of the field potentials after stimulation of the molecular layer of the dentate gyrus in slices of entorhinal lesioned rats suggests that even 55–65 days after the lesion, sprouting fibers have not restored normal electrophysiological properties in that area, but, rather, have produced an imbalance between excitation and inhibition (Clusmann et al., 1994), favoring the concept of a permanently altered circuitry. In a broader context, based on previous data from our laboratory, we are inclined to believe that lack of functional recovery after such hippocampal lesions may, possibly, be more the rule than the exception (Beck et al., 1990a, b ; Wree et al., 1993). Lesions of the dentate gyrus by ibotenic acid injections led to profoundly reduced LCGU in all major hippocampal areas up to 3 months postlesion, the only difference being that the contralateral hemisphere was as heavily affected as the ipsilateral (Wree et al., 1993). Previous work points to the involvement of the contralateral hemisphere also after entorhinal lesions. Increases in GFAP mRNA and GFAP protein were reported in the contralateral hemisphere after unilateral entorhinal lesions (Steward et al., 1990, 1993) and, similarly, densities of excitatory amino acids are also increased in the nondenervated hemisphere, possibly as a compensatory mechanism for the lost excitatory input (Ułas et al., 1990a, b ). Since the present data show only a short-term reduction of LCGU in the contralateral hemisphere and hardly any changes 3 months after the de-entorhination, the contralateral hemisphere apparently does recover from the initial depression rather comprehensively. As the entorhinal cortex projects primarily to the ipsilateral hippocampus and the contralateral projection is rather sparse and amounts to <5%, our data are fully compatible with the morphological findings. The idea has been advanced that the sprouted input from the contralateral entorhinal cortex might be especially valuable due to its homologous nature helping to compensate for the functional deficit, but since functional recovery does not take place within 3 months after the lesion, this hypothesis does not seem to be substantiated (Cotman et al., 1990).
It has been suggested that the entorhinal cortex plays an important role in learning and memory. Earlier work proposed that the process of reinnervation after unilateral entorhinal cortex lesions paralleled the recovery of alternation behavior in the rat (Loesche and Steward, 1977). However, this Suggestion was inferred from the remarkable coincidence of morphological findings with behavioral recovery and from the fact that after bilateral entorhinal lesions, behavioral recovery was absent. This view has been challenged by using postlesion halothane exposure, which suppresses reactive synaptogenesis. Surprisingly, recovery of spatial performance occurred in halothane-treated rats as well as in untreated controls, suggesting that behavioral recovery is not, in fact, due to reinnervation of the dentate but linked to other neuronal circuits (Levin et al., 1991). Although this conclusion would be compatible with the data of the present report, it seems advisable to take into account the complexity of behavioral studies. For example, retrograde memory retention scores after entorhinal lesions in mice were shown to depend heavily on the learning-surgery interval (Cho et al., 1993), a fact that doubtless impairs the comparison of experimental results.
Various studies have concentrated on altered receptor densities, and it appears that deafferentation of the hippocampus—whether due to entorhinal cortex ablation or lesions of the medial septum—have a long-lasting influence on kainate, or NMDA-or muscarinic receptors (Ułas et al., 1990a, b ; Harrell et al., 1994). The situation is less clear regarding GABA-receptors due to the invariable tissue shrinkage of the dentate molecular layer of up to 52% after removal of the entorhinal input (Goldowitz et al., 1982; Wagner et al., 1983). However, a staining for a PV-ir subset of GABAergic neurons has been intensively evaluated after entorhinal cortex ablation. These PV-ir neurons form part of the GABAergic inhibitory system within the hippocampus, controlling the excitatory input from the entorhinal cortex by virtue of their termination on cell bodies and axon-initial segments of pyramidal and granule cells (Kosaka et al., 1987; Lubbers and Frotscher, 1987; Nitsch et al., 1990; Soriano et al., 1990). It is intriguing to note that these neurons are not lost after the entorhinal ablation; rather, in the periphery of their dendritic fields, swellings and retractions occur along with indentations of the otherwise smooth dendrites on the ultrastructural level (Nitsch and Frotscher, 1993). Interestingly, these morphological alterations seem to progress gradually, show a chronic profile, and are mediated via NMDA-receptors (Nitsch and Frotscher, 1992). Even 55 days after entorhinal ablation, degenerating axon terminals occurred and dendrites were still reduced in the former termination zone of the perforant path 1 year after the lesion (Nitsch and Frotscher, 1993). These observations have a direct bearing on our findings and may be regarded as the morphological correlate of the irreversible reductions of glucose utilization within 3 months observed here.
Taken together, previous experimental evidence demonstrated that entorhinal lesions induce sprouting in the hippocampus associated with chronic morphological and electrophysiological alterations. The present report suggests that these repair mechanisms produce a neuronal network in disarray with permanently impaired functional properties. The findings again underscore the pivotal position of the entorhinal cortex in controlling hippocampal input and output. Considering that neuropathological alterations in the entorhinal cortex occur at a very early, clinically silent stage of Alzheimer's disease (Braak and Braak, 1991), the present data may also further the understanding of the chronic irreversible progression of this neurodegenerative disease.
Footnotes
Acknowledgment:
Thanks are due to K.-H- Augstein and R. Link for skillful technical support and G. Ritschel for art work. This work was supported by a grant from the Deutsche Forschungsgemeinschaft.